1957: First Results From Framingham Reported
The first findings in the Framingham Heart Study (FHS) were reported nine years after the study had been initiated as a result of President Truman signing the U.S. National Heart Act into law. I’ve presented some of those findings in an earlier column (1); here I review the important messages from that study.
In fact, the truly important messages from the FHS relate to what it did not find. Let’s review two critically important negative findings:
1. No dietary factor could explain the blood cholesterol concentrations measured in the entire cohort.
Recall that, unlike the dietary data in the Seven Countries Study, the dietary data collection in the FHS was meticulous (1). Thus, the findings are incontestable.
This first negative finding is incompatible with the diet-heart hypothesis, which holds that dietary factors, saturated fat in particular, increase blood cholesterol concentrations that are the direct cause of coronary heart disease (CHD).
T. J. Moore observes: “There is a considerable range of serum cholesterol levels within the Framingham Study Group. Something explains this inter-individual variation, but it is not diet (as measured here). Clearly if there is to be an attempt to manipulate blood cholesterol level in the general population, it would be desirable to know what these powerful but unspecified forces are” (2, p. 37; 3, p. 27).
As D. R. Jacobs et al. note, “Zero or near zero correlations were found between the various components of the diet and serum cholesterol levels” (4, p. 77).
Importantly, when the diets of men with either low or high blood cholesterol concentrations were compared, they “differed not at all in the amount or type of fat consumed,” Gary Taubes explains (3, p. 27).
George Mann claims, “The data clearly showed no relationship between dietary intakes of either fat or carbohydrate and the subjects’ level of cholesterolemia or their experience of CHD” (5, p. 9, my emphasis). This finding further destroys the diet-heart hypothesis.
William Castelli, who was the director of the FHS, would write many years after the initial results were published — in a medical journal that was not widely read: “In Framingham … we found that the people who ate the most cholesterol ate the most saturated fat, ate the most calories, weighed the least and were the most physically active” (6, p. 1371).
Interestingly, the article was merely an editorial written in response to the finding of an association (which cannot prove causation) between frequent nut consumption and a reduced risk of CHD (7). Without the need to provide substance to this editorial, perhaps Castelli would never have admitted what he wrote then. I am reminded of the belief that the guilty will usually admit to their guilt, if only obtusely.
Castelli continued:
Most of what we know about the effects of diet factors, particularly the saturation of fat and cholesterol, on blood lipid parameters derives from ward-type studies (8, 9). Alas, such findings, within a cohort studied over time have been disappointing (note confirmation bias — my addition), indeed the findings have been contradictory. For example, in Framingham, Mass, the more saturated fat one ate, the more cholesterol one ate, the more calories one ate, the lower the person’s serum cholesterol. The opposite of what one saw in the 26 metabolic ward studies, the opposite of what the equations provided by Hegsted et al. (8) and Keys et al. (9) would predict. Only the international comparisons showed that the world could be lined up on cholesterol intake or saturated fat intake, and it would correlate with the rate of CHD (10). (6, p. 1371)
What Castelli was actually saying is the following: In the FHS, diet determined neither the blood cholesterol concentration nor the risk of developing CHD. But this does not matter since other epidemiological studies — which, recall, cannot prove causation — have shown that as long as one investigates entire populations rather than individuals in the same population groups, there is a relationship between dietary fat intake and risk for developing CHD. Figure 1 exposes the subterfuge.
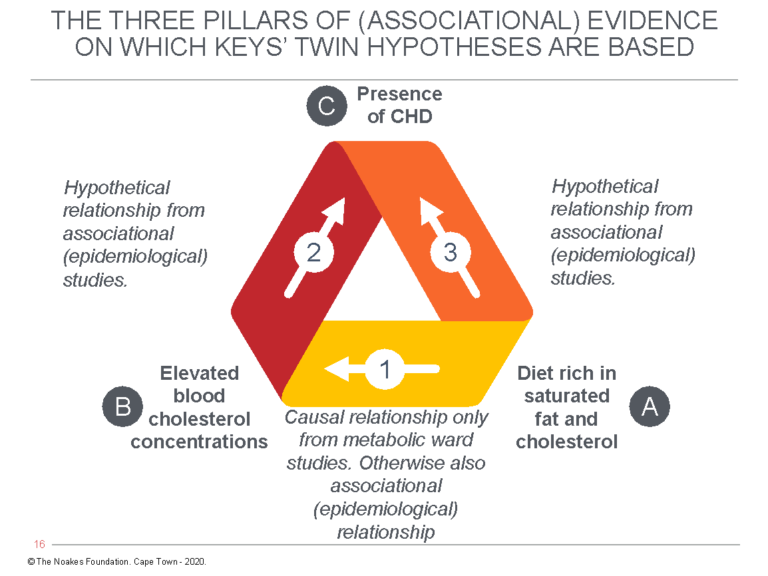
Figure 1: The diet-heart hypothesis predicts that a diet high in saturated fat and cholesterol (A) raises the blood cholesterol concentration (B), which is the direct cause of coronary heart disease (C). However, whereas the relationship between A → B can be detected through laboratory experimentation, the FHS sought to “prove” the relationships between B → C and A → C with an epidemiological observational study. For Ancel Keys’ diet-heart and lipid hypotheses to be true, the relationships shown by arrows 1, 2, and 3 must all be present at the same time in all the relevant studies, be they laboratory studies, epidemiological (associational) studies, or intervention trials (RCTs) in large populations. It’s not possible to have some of the relationships present in some studies but not in others and then to cherry-pick and choose those results that support the hypothesis as most relevant. This is what Keys and his acolytes have been doing for more than 67 years.
To show that he had at least learned something from the criticisms of Jacob Yerushalmy and Herbert Hilleboe (11, 12) about the weaknesses of epidemiological studies (arrows 2 and 3 in Figure 1), Castelli continued: “Of course, since these countries differed in many other ways, the possibility that some unidentified factor might explain the rate of CHD loomed in one’s thoughts” (6, p. 1371).
But then Castelli introduces his deception by arguing the absence of the necessary relationships (arrows 2 and 3 in Figure 1) in the FHS are immaterial since other studies have allegedly provided that required evidence: “Eventually, diet intervention trials were done, and where the follow-up got out beyond 3 years, they all show the same thing. The larger the percentage fall in cholesterol, the larger the percentage fall in CHD (13)” (6, p. 1371).
This response is disingenuous in the extreme, not just because it tries to deflect attention from the proofs required in Figure 1 if the statement is to be “true” and which the FHS was unable to provide, but more importantly because it is simply untrue.
The original paper (13) Castelli quotes does not provide any evidence from intervention trials showing “the larger the percentage fall in cholesterol, the larger the percentage fall in CHD.” The four studies to which Castelli refers were simply observational studies, as Jeremiah Stamler and Richard Shekelle acknowledge in the title of their article, “Dietary cholesterol and human coronary heart disease: The epidemiological evidence” (13). Perhaps Castelli did not ever understand the difference between observational studies and intervention trials or, perhaps, the reasons why only intervention trials can begin to identify causative factors (because they remove residual confounders). Or perhaps Castelli willfully chose to mislead the casual readers of his editorial. Either way, his blatant error should have been exposed by rigorous peer review.
However, even if the paper had been peer-reviewed, the “expert” reviewer likely would have been one of Castelli and Keys’ best mates, and it would not have been in his (since all were males) best interest to ask any awkward questions.
In effect, Castelli told a blatant fib. But Castelli was never one to get bogged down in complex detail. His understanding of the mechanisms of heart attack and its relation to diet was as simplistic as it could possibly be: “We now believe it’s the newest lipid deposits — where that greasy cheeseburger you just ate landed — that rupture and precipitate the majority of heart attacks” (14, p. 4). That the “grease” from the recent cheeseburger does not travel directly to be deposited in the coronary artery plaque was perhaps beyond the reach of Castelli’s understanding of metabolism. (The “grease” from the recent cheeseburger is distributed to the body’s fat stores, not to the linings of the coronary arteries).
The point is, as Taubes (3) and Nina Teicholz (15) have repeatedly warned, that when the evidence disproved Keys’ hypotheses, he and his acolytes simply ignored it. Or they labeled such studies as outliers that had to be wrong.
For example, no one ever remembers that in the FHS, half the persons who developed heart attacks had blood cholesterol concentrations below the normal level of 220 mg/dL (5.7 mmol/L) (15, p. 65; 16).
Many also often forget that those whose blood cholesterol concentrations fell progressively had a higher mortality rate.
Castelli et al. write: “There is a direct association between falling cholesterol levels over the 14 years (of the study) and mortality over the following 18 years (so that there was an) [11% overall and 14% CVD death rate increase per 1 mg/dL per year drop in cholesterol levels]” (16, p. 2176, my emphasis).
“After age 50 years there is no increased overall mortality from either high or low serum cholesterol levels,” they continue (16, p. 2176).
They also write that women, particularly those below the age of 64, can safely have blood cholesterol concentration of up to 294 mg/dL (7.6 mmol/L) without any increased risk of CHD (17). Apparently the sole concern of the male researchers in the FHS was the development of CHD in men; when women were found not to fit the male pattern, their different response was essentially ignored.
The same can be said of conditions other than those of the cardiovascular and cerebrovascular systems. For example, the fact that there was an inverse relationship between blood cholesterol concentrations and rates of colon cancer in men in the FHS has been largely ignored (18) (Figure 2 ).
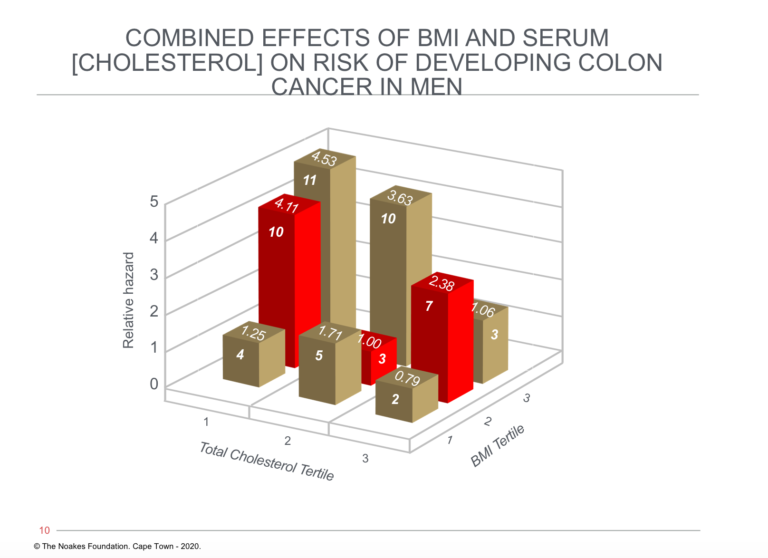
Figure 2: The relationship between blood cholesterol concentration, body mass index (BMI), and relative hazard for developing colon cancer. Note that risk of developing colon cancer is greatest in those with the highest BMIs and lowest blood cholesterol concentrations (tallest column on left). However, risk for those with the highest BMIs (BMI tertile 3, row of columns third from front) falls with increasing blood cholesterol concentrations, reaching an average risk (1.06) in those with the highest BMIs and highest blood cholesterol concentrations. Reproduced from Figure 3 in reference 18.
The remarkable observation related to Figure 2 is that a high blood cholesterol concentration was protective against colon cancer in those with elevated BMIs in the clinically obese range.
Thus, those men with the highest BMIs and highest blood cholesterol concentrations (final column on the extreme right of the figure) were at no greater risk of developing colon cancer than were the leanest men with the lowest blood cholesterol concentrations (shortest column on extreme left of the figure).
In women, there was no relationship between blood cholesterol concentrations and risk of colon cancer.
The authors conclude, “In men in the Framingham study, an inverse relation exists between baseline serum cholesterol levels and the incidence of colon cancer, even after the first 10 years of follow-up are eliminated. The especially higher risk among obese people with low cholesterol levels warrants additional attention” (18, p. 1042).
The reality is that all humans must die of something. But the singular focus on the effects of dietary change on blood cholesterol concentrations and CHD risk has left serious gaps in our understanding of how different diets affect health and risk of other diseases.
So there it is. If diet did not determine blood cholesterol concentrations in the FHS cohort, then according to the diet-heart hypothesis, diet could not be the cause of CHD in this population. End of story — regardless of what Stamler and Shekelle or anyone else might want us to believe.
In the end, Castelli conceded that the FHS had failed to prove that diet determined the blood cholesterol concentration in the free-living population in Framingham. His excuse was that the FHS was simply an epidemiological study and epidemiological studies are apparently flawed: “Epidemiologic data on diet is a risky analysis because it seems that people who eat the most cholesterol and the most fat have the lowest blood cholesterol levels. People who eat the most calories seem to weigh less than those who eat the least. This is just the opposite of what one might expect” (19, p. 58).
When another Keys acolyte, Scott Grundy, MD, wrote a final review (20) of the FHS findings, he predictably overstated the meagre positive outcomes — for example, the (poor) predictive value of blood cholesterol or LDL-cholesterol concentrations for future CHD risk — and he ignored all the inconvenient findings — for example, the absence of any links between dietary fat intake and either blood cholesterol concentrations or risk of CHD.
Those facts did not fit his evolving (false) narrative, so they safely could be ignored. His paternalistic attitude was clearly that the general public should not be confused with any inconvenient facts. Much better the public should simply be told what they must believe. That will spare them from having to think for themselves. This attitude of control would be essential if tens of millions were to be coerced into taking statin drugs in the false hope that statin use might protect them from future CHD. Grundy remained on the front lines (1, 21) promoting that false belief, all the while, I would guess, enjoying handsome rewards for his “commitment” to the diet-heart cause.
So once more, it’s not the theory that could ever be wrong. It’s just the fallible experiment that stubbornly refuses to come up with all the right answers.
2. There was no relationship between diet and risk of CHD.
As I described in a previous column (1), a careful dietary analysis of 1,000 participants in the FHS found “no relationship between dietary intakes of either fat or cholesterol … and [subjects’] experience of CHD” (5, p. 9). This was first reported in 1970 by the Framingham researchers in their carefully hidden document (1): “There is, in short, no suggestion of any relationship between diet and the subsequent development of CHD in the study group” (2, p. 37; 22).
Why is this finding so very important? Well because, as Taubes has argued in detail (3, p. 29), this absence of any relationship between diet and blood cholesterol concentrations or CHD risk has been found so often that it cannot continue to be ignored simply because it disproves what Keys and his team have brainwashed us to believe over the past 63 years. Here follows a brief review of much of that inconvenient evidence.
A study published in 1959 (23) reported that from a population of 106,000 persons living in northeastern North Dakota, the dietary intakes of 162 persons who developed CHD during one year was not different in any measured variable from the diet of 324 control subjects who remained healthy during that year. Importantly, both groups ate the same total number of daily calories and the same amounts of dietary fat — saturated fat, animal fat, and polyunsaturated fats, including linoleic acid.
The authors concluded: “Comparison of recent dietary histories of cases and controls revealed no differences in mean caloric intake, total fat consumption, or other major dietary constituents” (p. 1639). Significantly, lower incidences of CHD occurred in farmers compared to other occupations, in non-smokers, and in those who were more physically active.
The next inconvenient report came from the Framingham dietary data published in 1962 (24). Because the study had found no relationship between diet and blood cholesterol concentrations, it ultimately had to be hidden in the basements of the NIH offices in Washington, D.C. (1; 15, p. 67).
Next came the first report from the Western Electric Study, published in 1963. The study found there was no difference in dietary fat intake between persons who did or did not develop CHD after the first four and a half years of follow-up (25), even though blood cholesterol concentrations were higher in those who developed CHD. At the time of the study, the group considered to be eating a low-fat diet derived 39% of their calories from fat; the high-fat group, 45%. When the population was restudied 20 years later, it was found that “the amount of saturated fatty acids in the diet was not significantly associated with the risk of [CHD] death” (26).
In the same year, Jeremy Morris and colleagues (27) published a study on diet and plasma cholesterol concentrations in 99 bank employees in London. The study found blood cholesterol concentrations were unrelated to dietary factors in these men. Morris et al. concluded:
No closer association is evident between what these men ate and their individual cholesterol levels: diet thus does not seem to account for the wide range in cholesterol values that was found. The range may be crucial to the chances of developing ischaemic heart disease, and other causes of it should be sought. (p. 576)
In 1964, E. W. Lowenstein, then working for the World Health Organization in Geneva, collected all the studies he could find describing the diets of healthy men free of heart disease (15, p. 56; 28). He found their dietary fat intakes varied widely, from 7% among vegetarian Benedictine monks to 60-65% in the carnivorous Sambura and Somali. Despite such high fat intakes, and especially saturated fat derived almost exclusively from milk, blood cholesterol levels in the African pastoralists were low. Lowenstein wrote:
Contrary to what might have been expected, the pastoral tribes had a moderately low to low cholesterol and an apparently low incidence of coronary ischemic heart disease … despite the fact that their fat intake was very high. Obviously there are other factors in the way of life of these people which counteract any possible effect their diet may have on thrombogenesis sufficiently to prevent the development of coronary heart disease. (28, p. 184)
The finding that other carnivorous African tribes, like the Inuit, do not have high blood cholesterol concentrations was detailed in an earlier column (29).
Lowenstein also wrote, “It appears that a high saturated fat intake, even as high as 60 to 65%, is not necessarily detrimental if people do not overeat and [do] lead a physically active life” (28, p. 182).
Completely ignored in this conventional interpretation is the possibility of reverse causation: that these African pastoralists are healthy and physically active precisely because they eat a high-fat diet that protects them from ill health, perhaps by increasing their resistance to infectious diseases.
In 1964, Clarke Stout et al. described what came to be known as the Roseto effect (30). Roseto was an exclusively Italian-American town in eastern Pennsylvania with, in the early 1960s, “a remarkably low rate of coronary heart disease among the living, despite the fact that the conventional risk factors were found to be at least as prevalent in Roseto as in the two control communities” (31). It was also noted, “Rosetans eat at least as much animal fat as the average American” (32). Stewart Wolf — one of the original study authors — and colleagues later wrote:
The oil intake of the Rosetans was relatively low in olive oil. They used a great deal more lard than the wives of the people in this room use. One of their favorite dishes was fried peppers. They would fry the peppers in lard and they are very good. Then you’d take a piece of Italian bread and rub it around in the gravy that is left and eat that and that’s delicious! The Rosetans were very poor when they came and they are much more prosperous now. They eat everything. I’ve had many dinners with Rosetan families. They usually have more than one type of meat. When I eat ham I cut the rim of fat off and don’t eat it, same way with roast beef. They cut right through and eat it all. We were very elaborate in our study of their diet because we had Ansel Keyes (sic) breathing down our necks. Incidentally some of you may have seen the quote from Ansel Keyes (sic) in the New York Times recently. He said we may have put too much emphasis on cholesterol. (31, p. 67)
Heart disease rates and mortality rose over the next 25 years as the population began to move away from its traditional (Mediterranean) “values of southern Italian villagers.” This included dietary changes that “occurred … in the direction of what the American Heart Association calls ‘prudent.’ There was less consumption of animal fat, not a great difference, and there was less smoking and … less exercise” (31, p. 66).
The authors made two substantive conclusions. First, they concluded the lower rates of heart disease in Roseto in the 1960s were due to the “greater social solidarity and homogeneity in Roseto,” without any evidence that coronary risk factors were lower than in those living in the neighboring communities with higher heart disease mortality rates (33, p. 1092). Second, they concluded social change over the next 50 years produced the “sharply increased rates of heart attack among men under the age of 65” (33, p. 1092). This change was not prevented by an increased adoption of the AHA “prudent” diet, with its reduction in saturated fat intake and, importantly, in smoking rates. The findings mirror those from the MRFIT study in which a reduction in smoking incidence was not linked to a greater reduction in CHD in persons eating the “prudent” diet (34).
It is as if changing to the “prudent,” low-fat diet negates the benefits of stopping smoking — or at least that is one possible explanation for this paradoxical finding.
Dr. S. L. Malhotra was the chief medical officer of Western Railway in Mumbai (formerly Bombay), India. From that office, he studied the rates of hospital admission of railway employees in different regions of India. He showed the incidence of acute heart attacks was seven times higher in the rice-eating Indians living in the south than among the Punjabis in the north, who ate eight to 19 times more fat, chiefly of animal origin, and about nine times more sugar (35).
Malhotra also noted that the types of fats ingested by the two populations differed: In the north, the preponderant fat was saturated fat mainly from milk and fermented milk products, whereas in the south, vegetable fats dominated, particularly groundnut oil. Despite this, southerners died, on average, 12 years earlier than the Indians from the north. Naturally, he concluded his data did “not fit the hypothesis that low ratios of polyunsaturated to saturated fatty acids in food, or even an excess of sugar in food, contribute to an increased incidence of ischaemic heart disease” (36, p. 903).
His advice was to “eat more fermented milk products, such as yogurt, yogurt sherbet, and butter,” since the fatty acids in these foodstuffs are of a shorter length than fats derived from vegetables eaten by “South Indians, who would have … a preponderance of long-chain, complex fatty acid triglycerides” (35, p. 343). He noted long-chain saturated fatty acids promote thrombosis, whereas short-chain saturated fatty acids produce no such effect (37).
Teicholz makes the insightful point that even though Malhotra’s studies were published in a world-leading medical journal, the British Heart Journal, his work is almost never cited (15, p. 55). It’s as if it does not exist. Yet its findings are profoundly relevant since they reflect the experience of the second-most populous nation on Earth. Malhotra’s findings mirror the findings of a lifetime of work on the Indian subcontinent by Sir Robert McCarrison (38; 39, pp. 348-50).
In another earlier study, Malhotra and N. S. Pathania (40) had reported Indian vegetarians are not immune from CHD: “We find no reason to believe that vegetarians are less predisposed to coronary disease” (p. 530). The natural assumption is that those eating vegetarian diets will have lower blood cholesterol concentrations and therefore be at lower risk for the development of atherosclerosis (if Keys’ hypothesis is correct).
The next inconvenient finding with regard to Keys’ hypothesis came in 1965, when the diets and lifestyles of 26 matched pairs of Caucasian males living in Evans County, Georgia, who had either low or high blood cholesterol concentrations, were studied to “examine the possible relationship of serum cholesterol levels to dietary constituents and exercise” (41, p. 238). No correlation was found between the blood cholesterol concentration and the intake of any of the nutrients that were studied, including animal fat, animal protein, saturated fat, carbohydrate, vegetable fat, and vegetable protein. There was, however, a significant inverse relationship between physical activity and blood cholesterol concentrations.
This latter finding is disconcerting since it could suggest that any relationship between low blood cholesterol concentrations and lower rates of CHD might not result from the effects of a low-fat, vegetarian diet. Instead, the lower concentrations may simply be a marker of a person who is healthy enough to be physically active. The link between higher levels of physical activity and lower CHD rates is fairly well established (42).
In 1957, the Cardiovascular Health Center in Albany, New York, started to collect dietary information from male civil servants between the ages of 39 and 55 (43). The emphasis in the dietary analysis was on the kinds and amounts of fats that were eaten. In 1967, researchers analyzing the dietary data for 1,514 men found that these civil servants ate an average of 124 grams of fat daily, 74% of which was animal fat. The key finding of the study was: “The extremes of fat intake were accompanied by the same average serum cholesterol values. Conversely, when grouped by extremes of serum cholesterol levels, no differences were found in nutrient and caloric intake, except for the percentage of calories from carbohydrate” (43, p. 383).
In 1968 and 1969, Irish researchers published studies of the diets of male (44) and female (45) patients with CHD. In neither group was the amount or type of fat eaten any different from the fats eaten by healthy control subjects.
A 1976 study of approximately 2,000 men and women in Tecumseh, Michigan, found no relationship between cholesterol and triglycerides and “quality, quantity or proportion of fat, carbohydrates or protein consumed in the 24-hr recall period” (46, p. 1384). The researchers wrote, “These findings suggest that serum cholesterol and triglyceride levels among Americans are more dependent on degree of adiposity than on frequency of consumption of fat, sugar, starch or alcohol” (47, p. 1948).
In 1977, a British study of 337 healthy middle-aged men who were followed for 10-20 years found dietary factors showed “as expected, little or nothing” (48, p. 1312) in determining blood lipid concentrations. Dietary fat, and especially dietary cholesterol intake, “showed no association at all” with CHD events (p. 1312).
The Puerto Rico Heart Health Program (49) evaluated the relationship of diet to subsequent CHD over six years in 8,218 urban and rural Puerto Rican men. It found urban men who ate less, and especially fewer calories from carbohydrates such as rice and legumes, were more likely to suffer heart attacks. This same relationship was present in rural men but was not statistically significant.
The differences in dietary fat and carbohydrate intakes between those with and without CHD were miniscule. Complex carbohydrate intakes between those with or without CHD differed by just 13 grams/day in the urban men and 4 grams/day in the rural men. Total fat intakes differed by 1 gram/day in urban men with or without heart disease and 5 grams/day in rural men with or without heart disease.
The authors concluded that complex carbohydrates might be protective against CHD, which would mean Puerto Rico could rid itself of CHD if all its male citizens simply ate an extra 13 grams of starch a day, equivalent to one-half a cup of beans, lentils, or peas. They conveniently failed to point out that dietary fat intake in any form was unrelated to CHD risk.
The Honolulu Heart Study of 7,705 Japanese men living on the island of Oahu (50) also failed to find any significant differences in the types of dietary fat ingested by those who did or did not develop CHD over a six-year follow-up period. But persons who subsequently died from CHD ate 22 grams less of carbohydrate per day and 15 grams less of starch per day. They also ate fewer calories and drank less alcohol. The authors concluded, “There do not appear to be dietary differences between men remaining free of CHD and men going on to CHD death or MI (myocardial infarction) during a 6-year period of follow-up in this population of Japanese ancestry” (51, p. 1277).
Yet another analysis of these same data (52) found that cigarette smoking and hypertension made a more potent contribution to CHD risk in this population than did the blood cholesterol concentration. Glucose intolerance was also identified as a risk factor for fatal CHD.
The authors claimed the best advice for improving public health would be “either increasing carbohydrate intake or decreasing the percentage of calories from fat (and keeping caloric intake constant) … (as these result) in the same effect” (p. 675). They made this claim despite the evidence that the intake of calories from fat was unrelated to CHD mortality and that glucose intolerance was a risk factor for fatal CHD. Thus, the advice would cause harm in those subjects with “glucose intolerance” (i.e., insulin resistance).
Clearly, the authors were writing according to a script — the only acceptable script if they wanted their work to be published and acknowledged under the reign of the Keysian claim that CHD is exclusively caused by a diet too high in fat. Thus, they had to make the goal to replace dietary fat with carbohydrate regardless of the unforeseen consequences.
The Zutphen Study (53), which provided data for Keys’ Seven Countries Study, inconveniently found the 30 persons who died during a 10-year follow-up study of 857 men ate fewer calories, less animal protein, less vegetable protein, less saturated fat, less monounsaturated fat, less carbohydrate, and drank less alcohol. But they ate the same amount of dietary cholesterol, polyunsaturated fats, and dietary fiber as did those who remained healthy during this period.
The authors predictably concluded: “These results suggest that in a free-living population of middle-aged men, energy intake per kg of body weight is a stronger determinant of coronary heart disease than each individual macronutrient” (53). This conveniently allowed the authors to avoid mentioning the clear evidence that those who developed CHD ate less fat and less cholesterol than did those who remained free of CHD — more evidence disproving the diet-heart hypothesis.
D. L. McGee and colleagues further explored the data from the Honolulu Heart Program (54). They asked the until then forbidden question: What about deaths from diseases other than CHD? What is the effect of a low-fat diet on mortality from those diseases that have been ignored by Keys and his clan because of his obsession with diet and CHD?
Most inconveniently, the authors discovered “the consumption of dietary fat … is related inversely and significantly to total mortality” (54, p. 97, my emphasis) (Figure 3). Thus, whereas there were no significant relationships between grams of dietary fat, grams of dietary saturated fat, or grams of dietary cholesterol and “any of the specific causes of death examined” (p. 97), percentage of calories as dietary fat was inversely related to cancer (Figure 4) and stroke mortality (Figure 5), and directly related to CHD mortality. Specific causes of death included cancer, CHD, stroke, accidents and violence, and other causes.
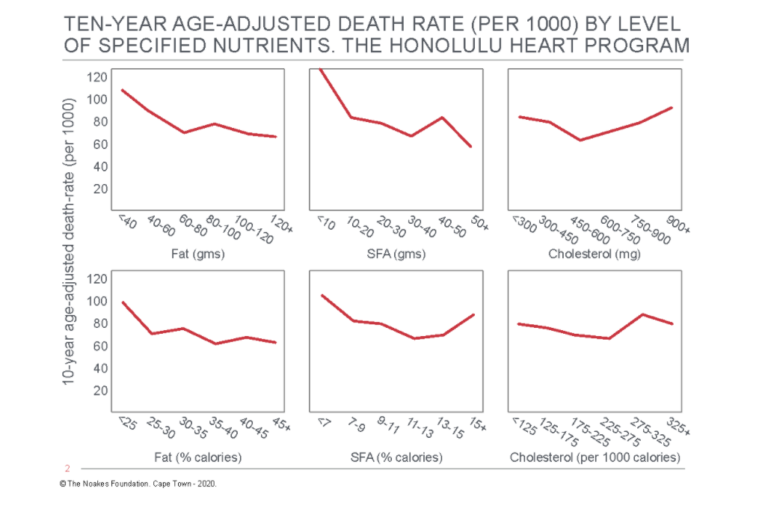
Figure 3: Age-adjusted death rate for total (all-cause) mortality in participants in the Honolulu Heart Program according to the intake of dietary fat, saturated fatty acids (SFA), and cholesterol. Note there is an inverse relationship between total mortality and total and SFA intake (columns on the left and in the center). Reproduced from reference 54, p. 99.
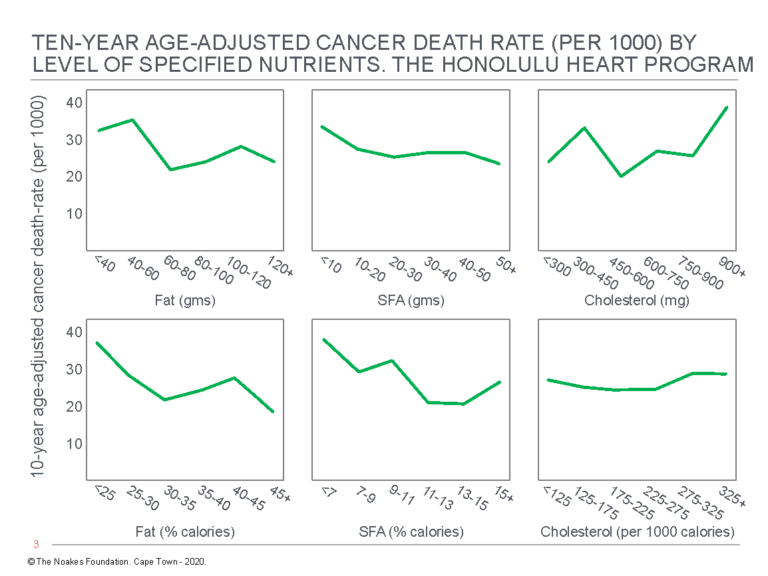
Figure 4: Age-adjusted death rate for cancer in participants in the Honolulu Heart Program by level of intake of dietary fat, SFA, and cholesterol. Note there is an inverse relationship between total cancer death rate and %fat and %SFA intake (columns on the left and in the center). Reproduced from reference 54, p. 100
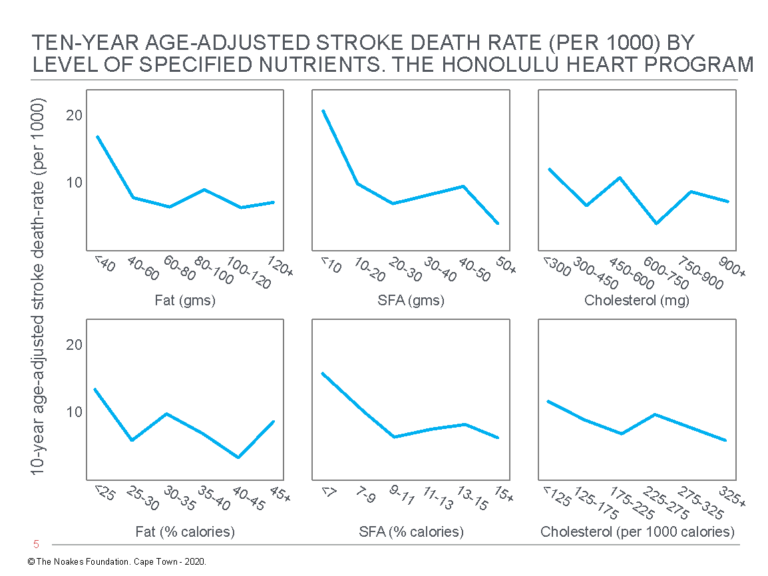
Figure 5: Age-adjusted stroke death rate in participants in the Honolulu Heart Program according to the intake of fat, saturated fatty acids (SFA), and cholesterol. Note there is an inverse relationship between stroke death rate and dietary %total fat and %SFA intake (columns on the left and in the center). Reproduced from reference 54, p. 102.
The authors were forced to conclude:
The overall results of this analysis present a dilemma. On the one hand, we are able to demonstrate a positive relationship between dietary intake of fat and cholesterol and CHD death. This relationship is believed to be a major cause of CHD rates in Western countries. On the other hand, we find that a low fat diet is associated with increased mortality from other causes, particularly mortality from cancer and from stroke, indicating no overall benefit from a low fat diet. (54, p. 104)
(Notably absent from their conclusion was even the vaguest suggestion that a low-fat diet might be the direct cause of high rates of cancer and stroke in Western countries).
The Israeli Male Civil Servants and Municipal Employees Study (55), originally termed the Israel Ischemic Heart Disease Study (56), quantified the food intakes and measured serum cholesterol concentrations in 10,000 male civil servants and government employees in Tel Aviv, Haifa, and Jerusalem in 1968. The authors reported there was “essentially no relationship between dietary elements and cholesterol” (56, p. 1126) and noted that two previous studies had essentially the same findings (24, 41). They also noted that this negative finding could not be attributed to studying too small a population or too little variation in what the civil servants were eating since this was a study of 10,000 individuals and “the variation among individuals in dietary factors is substantial” (56, p. 1126).
The authors posed the question, as had Framingham’s Castelli (6): Why do ward-based studies always find dietary changes produce predictable and statistically significant changes in blood cholesterol concentrations whereas population studies never find dietary fat intake determines the blood cholesterol concentration?
Their insightful explanation is, for obvious reasons, not frequently cited:
We suggest that the data are compatible with individual serum cholesterol values being dependent on several factors, perhaps diet, heredity, physical activity, and other factors and a random component with the dietary factor being an important but small component of the total (although not small with respect to the random component). If this is correct, we should observe, as we do, a very high correlation between dietary changes and serum cholesterol changes within individuals where all of the nondietary factors are presumably constant, and negligible correlation between an individual’s usual diet and his serum cholesterol value where the variation among individuals in the nondietary factors can be very great. One corollary of the diet-cholesterol relationship as pictured above is that international comparisons of cholesterol levels in relation to estimated international dietary differences may be suspect. If dietary elements are but one among several factors related to cholesterol levels and, furthermore, if they do not exert a controlling or overriding influence on cholesterol level, then the average cholesterol level in a community is likely to be more a function of nondietary factors than it is of dietary elements. Under these conditions, the difference in cholesterol levels among communities are also likely to be more related to nondietary than to dietary factors. (56, p. 1126, my emphasis)
Is it not possible that those same nondietary factors might also explain differences in heart disease rates incorrectly attributed to differences in dietary fat intakes by Ancel Keys?
The 23-year follow-up study of this population found that, compared to smoking, hypertension, and T2DM, blood cholesterol concentrations made a small contribution to total CHD events and CHD mortality (55). But the strongest risk factor was the one that would continue to be ignored because it runs contrary to the accepted dogma. As the authors of the follow-up study explained, “Diabetes mellitus therefore remained the strongest precursor of long-term coronary death in CHD-free individuals in this cohort of middle-aged and elderly men” (55, p. 112)
The authors problematically also wrote: “The absence of an independent contribution of dietary variables to CHD risk is of secondary importance given the established effect of diets replacing saturated fat by other fat types on the blood lipid profile” (55, p. 119). In other words, they seemed to know that replacing dietary saturated fat with polyunsaturated fats must reduce CHD risk since this dietary change lowers blood cholesterol concentrations. They then, however, cited evidence from their country that directly conflicted with these assumptions.
First, Israel had one of the very highest rates of polyunsaturated fat consumption in the world (58, 59). If this dietary choice was protective, the study might have detected it. Indeed, as the authors of a later analysis explained, “In fact, Israeli Jews may be regarded as a population-based dietary experiment of the effect of a high omega-6 polyunsaturated fatty acid (PUFA) diet, a diet that until recently was widely recommended” (59, p. 1134).
Unfortunately, this fascination with a high-PUFA diet has not gone well for the Israelis. In fact, it has produced the “Israeli paradox,” so called because Israel has a “high prevalence of cardiovascular diseases, hypertension, non-insulin dependent diabetes mellitus and obesity — all diseases that are associated with hyperinsulinemia (HI) and insulin resistance (IR) … [and] also an increased cancer incidence and mortality rate especially in women, compared with western countries” (p. 1134). These findings are paradoxical if PUFAs are protective against heart disease and cancer.
Instead, Daniel Yam et al. question whether, “rather than being beneficial, high omega-6 PUFA diets may have some long term side effects” (59, p. 1134).
Second, mortality among Israelis fell between 1974 and 1979 (60) in the absence of any overt lifestyle modifications, including unchanged blood cholesterol concentrations and smoking habits, and with dietary changes that are the opposite of those that were promoted. These changes included an increase in the percentage of daily energy derived from fats and a reduction in carbohydrate, notably starch, consumption. As U. Goldbourt and H. N. Neufeld explain, “Changes in CHD mortality were not accompanied by putative anti-atherogenic trends in eating habits” (60, p. 63).
Unless, of course, Keys’ anti-atherogenic PUFA diet is actually pro-atherogenic.
A 1985 re-analysis (61) of an earlier 1964 study of Irish siblings living in either Dublin or Boston (62) found brothers who reported eating more saturated fat and less polyunsaturated fat in the early 1960s had slightly higher rates of CHD. But the senior author who worked in Keys’ department at the University of Minnesota was surprisingly restrained in his conclusions: “Overall, these results tend to support the hypothesis that diet is related, albeit weakly, to the development of coronary heart disease” (62, p. 811).
So how did Keys and his acolytes explain away all these most inconvenient findings that were clearly incongruent with the diet-heart hypothesis?
Two of Keys’ most trusted lieutenants — Stamler, who had directed the MRFIT study, and Shekelle — discounted the inconvenient findings from the Western Electric Study with this rationalization:
If viewed in isolation, the conclusions that can be drawn from a single epidemiologic study are limited. Within the context of the total literature, however, the present observations support the conclusion that the (fat) content of the diet affects the level of serum cholesterol and the long term risk of death from CHD in middle-aged American men. (26)
Yet the analysis presented here shows the habitual diet eaten by those with CHD has never been shown to be any different from those without the disease. Thus, dietary fat intake cannot be the single factor determining who will develop CHD and who will be spared.
In essence, like all the past and present members of Keys’ coalition, Shekelle and Stamler confirmed that it matters not what the evidence shows, because only they are privileged to comprehend a divine truth.
Contrast that to the statement of a real scientist, Albert Einstein, who observed it matters not how much evidence supports a theory — at any moment a single study can disprove it.
Keys’ explanations in defense of his hypothesis became ever more obtuse. This time he blamed the experimental subjects. Clearly they were the cause of the “problem”:
Within a culturally homogenous population it is fruitless to attempt to characterize with any reliability the individuals in respect to the nutrient variables relevant to serum lipids, atherosclerosis and coronary heart disease. The conclusion refers to methods used so far, but the methods are not responsible; the inescapable limitation is the spontaneous variability of the individuals themselves. (63, pp. 1155-1156)
What Keys was effectively arguing is that individuals in any single “culture” differ so greatly in how they respond to what they eat that it makes it impossible to detect relationships between anything in their diet and their blood lipid concentrations or their risk of developing atherosclerosis or CHD.
Stated differently, he was arguing that we know diet alters blood lipid values and the risk of developing atherosclerosis or CHD, so if we can’t identify those causative factors in individuals living similar lives in the same cultural setting and eating similar foods, the problem lies with the individuals. They simply don’t do what we expect them to do.
Nevertheless, some of those “spontaneously variable” individuals die of heart disease and others do not. Despite all his efforts over many decades, Keys and his researchers were unable to find a single dietary factor that might explain this difference (64).
In summary, the FHS effectively disproved the diet-heart hypothesis by showing blood cholesterol concentrations in the free-living population were unrelated to what people ate, particularly the amount and type of fat in the diet. Unlike Keys’ Seven Countries Study (15, pp. 39-42; 65), the dietary analysis was very carefully conducted and was a true representation of what the cohort was eating. That the Keys acolytes ensured the data was buried deep in the vaults of the NHLBI archives in Washington, D.C., confirms just how potentially explosive those data were (15, pp. 66-67).
I have pondered for many hours how Keys, his contemporary acolytes, and his modern legion of academic devotees managed to get away without ever exposing these truths to the general public.
The reason, I believe, is that the FHS did show that an elevated blood cholesterol concentration is a predictor, albeit relatively weak, of CHD risk (Figure 1 in reference 1), but the risk has always been overstated, as Grundy classically did with the MRFIT data (20; Figures 4 and 5 in reference 1). So what is the truth?
Figure 6 shows the 20-year FHS data as reported by Moore (2, p. 35).
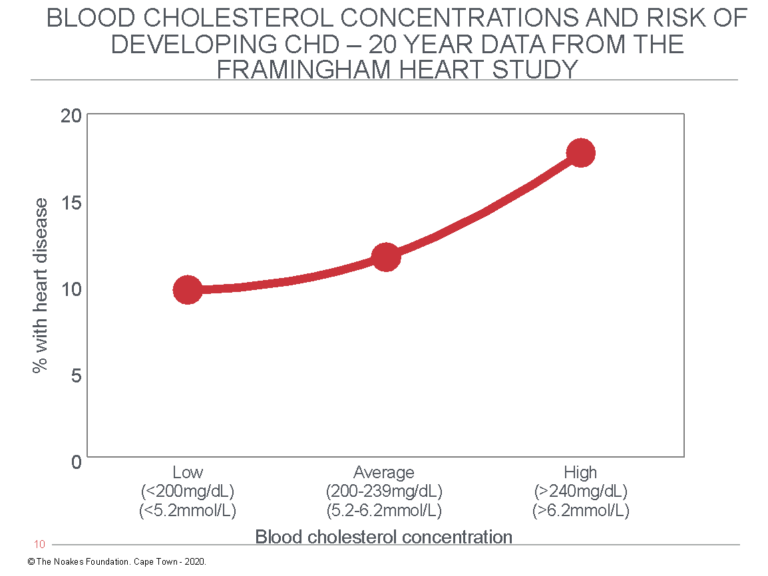
Figure 6: Redrawn from the FHS data reported by Moore (2, p. 35), this graph shows that the percentage of study subjects who developed CHD during 20 years of observation increased with rising blood cholesterol concentrations. But the increase (8%) associated with higher blood cholesterol levels was less than the prevalence of CHD in the group with the lowest blood cholesterol concentrations (10%). In other words, factors other than the blood cholesterol concentration explained the presence of CHD even in those with the lowest blood cholesterol concentrations. Other studies from the same population reported higher blood HDL cholesterol concentrations in this population were better predictors of reduced CHD risk and that “at all ages total cholesterol was not associated with the risk of coronary heart disease” (66, p. 707).
Figure 6 shows that whereas 10% of subjects with blood cholesterol concentrations below 200 mg/dL (5.2 mmol/L) developed CHD during 20 years of study, CHD developed in 18% of those with blood cholesterol concentrations higher than 240 mg/dL (6.2 mmol/L). Thus, the difference in 20-year CHD rates between those with “healthy” blood cholesterol concentrations and those in whom blood cholesterol concentrations were “dangerously elevated” was eight cases per 100 persons at risk per 20 years, or 0.4 persons per 100 per year. While this difference is “statistically significant” in scientific terms, in terms of what it means to humans with “elevated” blood cholesterol concentrations, it is utterly meaningless.
Stated differently, this graph shows the annual rate of new CHD events in the group with the lowest blood cholesterol concentrations was 0.5%, whereas in the group with the highest blood cholesterol concentrations it was 0.9%, a difference of 0.4%. This means that if the difference in CHD incidence is due only and exclusively to differences in blood cholesterol concentrations, one would need to treat 250 persons with “dangerously elevated” blood cholesterol concentrations for one person to benefit for each year of treatment.
But even without any treatment, at the end of 20 years, 90% of those with the lowest blood cholesterol concentrations would still be alive compared to 82% of those with the highest blood cholesterol concentrations. The question becomes: How is it possible to identify the small percentage of subjects who will die in the next 20 years, regardless of whether they have “high” or “low” blood cholesterol concentrations? And what would be the risk of artificially lowering blood cholesterol concentrations by whatever means if we know lower blood cholesterol concentrations are associated with increased mortality (16) and a greater risk of cancer, particularly colon cancer (18) — and perhaps many other adverse consequences still to be described (67)?
This poverty of LDL cholesterol as a predictor of CHD risk was clearly appreciated by the FHS researchers already in the early 1970s. In 1974, in a private communication to William Kannel, then Director of the Framingham Study Tavia Gordon wrote:
We have no proof that anyone would lower his chances of developing CHD by reducing his serum cholesterol level, whether that level be 220 or 320 or whatever. The truth is, however, that we would need many more observations than we have to be able to assert from experience that at very low levels an increment of risk with increased serum cholesterol really occurs. (68, p. 867)
It was perhaps not surprising that in 1977, Gordon and the FHS team dropped a colossal scientific bombshell when they announced that in the FHS population, HDL cholesterol predicted protection from CHD, whereas “at all ages total cholesterol was not associated with the risk of coronary heart disease” (66, p. 707, my emphasis; Figure 2 in reference 1).
In the same year, the same group reported “HDL cholesterol concentration is inversely related to subsequent development of CHD” (69, p. 771), and serum triglyceride concentrations also showed a “direct relationship” with the “prevalence of CHD” (69, pp. 771-772).
Naturally, this was not well received since it conflicted absolutely with the lipid hypothesis.
The result was that the maverick resolve of the FHS researchers lasted precisely two years, by which time they had issued a retraction of sorts.
In January of 1979, Kannel, Castelli, and Gordon were again singing the same tune; LDL cholesterol had been restored to its former position of dominance. Thus, they wrote serum total cholesterol shows “a powerful relation of this lipid to the subsequent development of coronary heart disease” (70, p. 85). They also claimed “prospective epidemiologic studies around the world (have documented) that risk of coronary heart disease is strongly related to serum total cholesterol within the general population” (p. 86). However, it seems they weren’t yet completely convinced: “For one thing, the strength of the association wanes progressively with advancing age so that serum cholesterol is no longer a predictor of risk in men beyond age 65” (p. 86).
In an editorial published 10 months later in the Lancet, however, they could afford to be less circumspect and more assertive in their conclusions:
There is really no doubt that the serum total cholesterol is related to the rate of development of clinical CHD both within and among populations. The association is dose-related, occurs for both sexes, precedes the disease, is independent of other risk factors, and can be demonstrated for all clinical manifestations of CHD with great consistency in diverse population samples all over the world. (71, p. 950)
This statement is untrue in many respects, not least because the risk of sudden coronary death was unrelated to the blood cholesterol concentration in the FHS population (72).
The authors concluded with an important concession:
It must be admitted that the results achieved in trials to reduce CHD risk by drugs or diet designed to lower the serum-total-cholesterol content have been disappointing. This may be a consequence of the small reductions in cholesterol achieved, coupled with the short duration of the trials, usually involving people with advanced lesions. (71, p. 951)
The only proper conclusion was that the failure of reductions in blood cholesterol concentrations to prevent progression of CHD indicates that the hypothesis on which the prediction is based is false. Naturally, this conclusion could never be entertained.
In time, the authors would appreciate that the combination of high blood triglyceride and low HDL-cholesterol concentrations occurs in persons with insulin resistance, and low(er) carbohydrate diets can reverse the hypertriglyceridemia (73).
Ultimately, this was all confirmed in the 14-year follow-up study of 2,910 of the offspring of the original FHS cohort (74). As reported previously, the study established that insulin resistance, elevated blood triglycerides, and low HDL-cholesterol concentrations are the best predictors of future CHD risk in this population (74; Figure 6 in reference 1).
This study also neatly explained why two of the key markers of insulin resistance — blood triglyceride and HDL-cholesterol concentrations — are superior predictors of CHD risk, and when other markers of insulin resistance are included, their predictive value increases.
More recently, the Framingham Offspring Study has produced another interesting finding. The consumption of sugar-sweetened beverages was associated with decreases in blood HDL cholesterol and increases in blood triglyceride concentrations (75). Presumably, these responses would be worsened in persons with insulin resistance.
How Keys and His Followers Hid the Inconvenient Findings of the FHS and SCS
One thing I think Keys and his acolytes did successfully over many years was to conflate the findings of the FHS with those from the SCS, thus effectively confusing members of the medical profession and general public.
What Keys et al. really found was that even if a higher blood cholesterol concentration predicted a slightly higher incidence of CHD in the FHS (Figure 6), this could not be explained by differences in diet, particularly by higher rates of saturated fat intake in those with CHD. They knew admitting that finding would destroy their hypothesis and end their dreams, so they hid the data (22). The world suffered as a result.
In discussing the release of the first reports of the SCS in 1970 (76), Keys also skillfully hid the evidence that if a high-fat diet was indeed causing heart disease, it was not acting through elevated blood cholesterol concentrations.
Thus, whereas there was a decent linear (associational) relationship between blood cholesterol concentrations and rates of CHD in the SCS cohorts (Figure 5 in reference 76), the relationship between the percentage of saturated fat in the diet and CHD rates was less impressive and perhaps rather more illusory (Figure 6 in reference 76).
Hidden deep in the SCS report was this obtuse statement:
For diet, within-individual variation in the same cohort was of the same order as between-individual variation. This provided another demonstration of statistical theory: under such conditions, individual serum cholesterol and nutrient composition of diet cannot be found correlated without repeated surveys to reduce the effects of intra-individual variation. (77, p. 162)
In other words, whatever the findings might be in different populations, in the individuals comprising those populations, there was no relationship between dietary factors, including saturated fat intake, and blood cholesterol concentrations.
This means Keys and his colleagues had provided the data to disprove their hypothesis — which is the goal of all good science — but because they were not good and honest scientists, they explained away or hid the relevant findings.
The remarkable point is that our natural inclination — the result of years of brainwashing — is to believe that an individual’s diet, in particular their saturated fat intake, determines their blood cholesterol concentration and hence their risk of CHD.
When we discover that essentially the innumerable efforts to prove this relationship have failed, it’s very, very hard to accept, because we must first acknowledge that we have been duped.
It is as Richard Feynman said: “The first principle is that you must not fool yourself. And you are the easiest to fool.”
Additional Reading
References
- Noakes TD. It’s the insulin resistance, stupid: Part 9. CrossFit.com. 11 December 2019. Available here.
- Moore TJ. The cholesterol myth. The Atlantic. 1989;264(September):37; Moore TJ. Heart Failure: A Critical Inquiry Into American Medicine and the Revolution in Heart Care. New York, NY: Simon and Schuster, 1989.
- Taubes G. Good Calories, Bad Calories: Fats, Carbs, and the Controversial Science of Diet and Health. Anchor Books, New York, NY. 2008.
- Jacobs DR, Anderson JT, Blackburn H. Diet and serum cholesterol. Do zero correlations negate the relationship? Am J Epidemiol 1979;110:77-87
- Mann GV. A short history of the diet/heart hypothesis. In: Coronary Heart Disease: The Dietary Sense and Nonsense. An Evaluation by Scientists. Mann GV, Ed. London, England: Janus Publishing Company, 1993. pp. 1-17.
- Castelli WP. Concerning the possibilities of a Nut… Arch Intern Med 1992;152:1371-1372.
- Fraser GE, Sabate J, Beeson WL, et al. A possible protective effect of nut consumption on risk of coronary heart disease. The Adventist Health Study. Arch Intern Med 1992;152:1416-1424.
- Hegsted DM, McGandy RB, Myer ML, et al. Quantitative effects of dietary fat on serum cholesterol in man. Am J Clin Nutr 1965;17:281-295.
- Keys A, Anderson JT, Grande F. Prediction of serum cholesterol responses of men to changes in fat in the diet. Lancet 1957;2:959-966.
- Keys A. Coronary heart disease in seven countries. Circulation 1970;41(suppl 1):1-211.
- Yerushalmy J, Hilleboe HE. Fat in the diet and mortality from heart disease. A Methodological note. New Y State J Med 1957:57:2343-2354.
- Noakes TD. It’s the insulin resistance, stupid: Part 7. CrossFit.com. 11 November 2019. Available here.
- Stamler J, Shekelle R. Dietary cholesterol and human coronary heart disease. The Epidemiological Evidence. Arch Path Lab Med 1988;112:1032-1040.
- Brody J. Scientist at work: William Castelli; preaching the gospel of healthy hearts. The New York Times February 8th 1994.
- Teicholz N. The Big Fat Surprise: Why Butter, Meat and Cheese Belong in a Heathy Diet. Simon and Schuster, New York, NY. 2014.
- Anderson KM, Castelli WP, Levy D. Cholesterol and mortality. 30 years of follow-up from the Framingham study. JAMA 1987;257:2176-2180.
- Kannel WB. Metabolic risk factors for coronary heart disease in women: Perspective from the Framingham Study. Am Heart J 1987:114:413-419.
- Kreger BE, Anderson KM, Schatzkin A, et al. Serum cholesterol level, body mass index, and the risk of colon cancer. The Framingham Study. Cancer 1992;70:1038-1043.
- Enig MG. Diet, serum cholesterol, and coronary heart disease. Chapter 3: In: Coronary Heart Disease: The Dietary Sense and Nonsense. An Evaluation by Scientists. Mann GV, Ed. Janus Publishing Company, London. 1993; pp.36-60
- Grundy SM, Balady GJ, Criqui MA, et al. AHA Scientific Statement. Primary prevention of coronary heart disease: Guidance from Framingham. A statement for healthcare professionals from the AHA Task Force on Risk Reduction. Circulation 1998;97:1876-1887.
- Grundy SM. Cholesterol and coronary heart disease. A new era. JAMA 1986;256:2849-2858.
- Anon. The Framingham Study: an Epidemiological Investigation of Cardiovascular Disease. DHEW Pub. No. (NIH) 74-618,74-478, 74-599,76-1083.
- Zukel WJ, Lewis RH, Enterline PE, et al. A short-term community study of the epidemiology of coronary heart disease. A preliminary report on the North Dakota Study. Am J Publ Health 1959;49:1630-1639.
- Kannel W, Dawber T, Glennon W, et al. Preliminary Report: the determinants and clinical significance of serum cholesterol. Mass J Med Technol 1962;4:11.
- Paul O, Lepper MJ, Phelan WH, et al. A longitudinal study of coronary heart disease. Circulation 1963;28;20-31.
- Shekelle RB, Shryock AM, Paul O, et al. Diet, serum cholesterol, and death from coronary heart disease – The Western Electric Study. N Eng J Med 1981;304:65-70.
- Morris JN, Marr JW, Heady JA, et al. Diet and plasma cholesterol in 99 Bank men. BMJ 1963;1:571-576.
- Lowenstein FW. Epidemiologic investigations in relation to diet in groups who show little atherosclerosis and are almost free of coronary ischemic heart disease. Am J Clin Nutr 1964;15:176-186.
- Noakes TD. Ancel Keys’ cholesterol con, part 4. CrossFit.com. 19 June 2020. Available here.
- Stout C, Morrow J, Brandt EN, et al. Unusually low incidence of death from myocardial infarction. Study of an Italian American community in Pennsylvania. JAMA 1964;188:845-849.
- Wolf S, Herrenkohl RC, Lasker J, et al. Roseto, Pennsylvania 25 years later – highlights of a medical and sociologic survey. Trans Am Clin Climatol Assoc 1988;100:57-67.
- Wolf S. Mortality from myocardial infarction in Roseto. JAMA 1966;195:142.
- Egolf B, Lasker J, Wolf S, et al. The Roseto Effect: A 50-year comparison of mortality rates. Am J Publ Health 1992;82:1089-1092.
- Noakes TD. Ancel Keys’ cholesterol con, part 9. Forthcoming on CrossFit.com.
- Malhotra SL. Geographical aspects of acute myocardial infarction in India with special reference to patterns of diet and eating. Brit Heart J 1967;29:337-344.
- Malhotra SL. Epidemiology of ischaemic heart disease in India with special reference to causation. Brit Heart J 1967;29:895-905.
- Connor WE, Poole JCF. The effect of fatty acids on the formation of thrombi. Q J Exp Physiol 1961;46:1-7.
- McCarrison R. Studies in deficiency disease. Andesite Press, 2015; McCarrison R. Nutrition in health and disease. BMJ 1936;2:611-615.
- Noakes TD, Sboros M. Nutrition on Trial. How the Diet Dictators Tried to Destroy a Top Scientist. Columbus Publishing Ltd, UK. 2019, pp.1-488.
- Malhotra RP, Pathania NS. Some aetiological aspects of coronary heart disease. An Indian point of view based on a study of 867 cases seen during 1948-1955. BMJ 1958;2:528-531.
- Stulb SC, McDonough JR, Greenberg BG, et al. The relationship of nutrient intake and exercise to serum cholesterol levels in white males in Evans County, Georgia. Am J Clin Nutr 1965;16:238-242.
- Paffenbarger RS, Hyde RT. Exercise in the prevention of coronary heart disease. Prev Med 1984;13:3-22.
- Browe JH, Morlley DM, Logrillo VM, et al. Diet and heart disease in the cardiovascular health center. J Am Diet Assoc 1967;50:376-384.
- Finegan A, Hickey N, Maurer B, et al. Diet and coronary heart disease: Dietary analysis on 100 male patients. Am J Clin Nutr 1968;21:143-148.
- Finegan A, Hickey N, Maurer B, et al. Diet and coronary heart disease: Dietary analysis on fifty females. Am J Clin Nutr 1969;22:8-9.
- Nichols AB, Ravenscroft C, Lamphiear DE, et al. Daily nutritional intake and serum lipid levels. The Tecumseh Study. Am J Clin Nutr 1976;29:1384-1392.
- Nichols AB, Ravenscroft C, Lamphiear DE, et al. Independence of serum lipid levels and dietary habits. The Tecumseh Study. JAMA 1976;236:1948-1953.
- Morris JN, Marr JW, Clayton DG. Diet and heart: a postscript. BMJ 1977;2:1307-1314.
- Garcia-Palmieri MR, Sorlie P, Tillotson J, et al. Relationship of dietary intake to subsequent coronary heart disease incidence. The Puerto Rico Heart Health Program. Am J Clin Nutr 1980;33:1818-1827.
- Yano K, Rhoads GG, Kagan A, et al. Dietary intake and the risk of coronary heart disease in Japanese men living in Hawaii. Am J Clin Nutr 1978;31:1270-1279.
- McGee DL, Reed DM, Yano K, et al. Ten-year incidence of coronary heart disease in the Honolulu Heart Program. Relation to nutrient intake. Am J Epidemiol 1984;119:667-676.
- Yano K, Reed DM, McGee DL. Ten-year incidence of coronary heart disease in the Honolulu Heart Program. Relationship to biologic and lifestyle characteristics. Am J Epidemiol 1984;119:653-666.
- Kromhout D, Coulander CDL. Diet, prevalence and 10-year mortality from coronary heart disease in 871 middle-aged men. The Zutpen Study. Am J Epidemiol 1984;119:733-741.
- McGee D, Reed D, Stemmerman G, et al. The relationship of dietary fat and cholesterol to mortality in 10 years: The Honolulu Heart Program. Int J Epidemiol 1985;14:97-105.
- Kahn HA, Medalie JH, Neufeld HN, et al. Serum cholesterol: Its distribution and association with dietary and other variables in a survey of 10,000 men. Isr J Med Sci 1969;5:1117-1127.
- Goldbourt U, Yaari S, Medalie JH. Factors predictive of long-term coronary heart disease mortality among 10,059 male Israeli civil servants and municipal employees. A 23-year mortality follow-up in the Israeli Ischemic Heart Disease Study. Cardiology 1993;82:100-121.
- Medalie JH, Kahn HA, Neufeld HN, et al. Five-year myocardial infarction incidence – II. Association of single variables to age and birthplace. J Clin Endocrinol 1973;26:329-349.
- Kaufman NA, Dennis BH, Heiss G, et al. Comparison of nutrient intakes of selected populations in the United States and Israel: the lipid research clinics prevalence study. Am J Clin Nutr 1986;43:604-620.
- Yam D, Eliraz A, Berry EM. Diet and disease – the Israeli Paradox: Possible dangers of high omega-6 polyunsaturated fatty acid diet. Isr J Med Sci 1996;32:1134-1143.
- Goldbourt U, Neufeld HN. Disease mortality and related factors in Israel. Cardiology 1985;72:63-74.
- Trulson MF, Clancy RE, Jessop WJE, et al. Comparisons of siblings in Boston and Ireland: Physical, biochemical, and dietary findings. J Am Diet Soc 1964;45:225-229.
- Kushi LH, Lew RA, Stare FJ, et al. Diet and 20-year mortality from coronary heart disease. The Ireland-Boston Diet-Heart Study. N Eng J Med 1985;312:811-818.
- Keys A. Dietary epidemiology. Am J Clin Nutr 1967;20:1151-1157.
- Gordon T, Kagan A, Garcia-Palmieri M, et al. Diet and its relation to coronary heart disease and death in three populations. Circulation 1981;63: 500-515.
- Noakes TD. It’s the insulin resistance, stupid: Part 8. CrossFit.com. 24 Nov. 2019. Available here.
- Gordon T, Castelli WP, Hjortland MC, et al. High density lipoprotein as a protective factor against coronary heart disease. The Framingham study. Am J Med 1977;62:707-714.
- Jacobs D, Blackburn H, Higgins M, et al. Report of the conference on low blood cholesterol:mortality associations. Circulation 1992;86:1046-1060.
- Reiser R. Oversimplification of diet:coronary heart disease relationship and exaggerated diet recommendations. Am J Clin Nutr 1978;31:865-875.
- Castelli WP, Doyle JT, Gordon T, et al. HDL cholesterol and other lipids in coronary heart disease. The Coorperative Lipoprotein Phenotyping Study. Circulation 1977;55:767-772.
- Kannel WB, Castelli WP, Gordon T. Cholesterol in the prediction of atherosclerotic disease: New perspectives from the Framingham Study. Ann Intern Med 1979;90;85-91.
- Kannel WB, Castelli WP. Is the serum cholesterol an anachronism? Lancet 1979;2:950-951.
- Kannel W, Thomas EH. Sudden Coronary Death. The Framingham Study. Ann NY Acad Sci 1982;382:3-21.
- Hulley SB, Burrows M, Wilson WS, et al. Lipid and lipoprotein responses of hypertriglyceridaemic outpatients to a low-carbohydrate modification of the A.H.A. fat-controlled diet. Lancet 1972;2:551-555.
- Robins SJ, Lyass A, Zachariah JP, et al. Insulin resistance and the relation of dyslipidemia to coronary heart disease. The Framingham Heart Study. Arterioscler Thromb Vasc Biol 2011;31:1208-1214.
- Haslam DE, Peloso GM, Herman MA, et al. Beverage consumption and longitudinal changes in lipoprotein concentrations and incidence dyslipidemia in US adults: The Framingham Heart Study. J Am Heart Assoc 2020;9:e014083.
- Noakes TD. Ancel Keys’ cholesterol con, part 7. Forthcoming on CrossFit.com.
- Blackburn H, Menotti A. Major results of the Seven Countries Study. Chapter 14. In: The Seven Countries Study. A Scientific Adventure in Cardiovascular Disease Epidemiology. Eds: Kromhout D, Menotti A, Blackburn H. Utrecht, The Netherlands. 1993;159-168.
- Leren P. Prevention of Coronary Heart Disease: Some Results from the Oslo Secondary and Primary Intervention Studies. J Am Coll Nutr 1989;5:407-410.
- Noakes TD. It’s the insulin resistance, stupid: Part 1. CrossFit.com. 7 July 2019. Available here.
Comments on Ancel Keys' Cholesterol Con, Part 5
The nations comparison has been done long ago http://hopefulgeranium.blogspot.com/2014/09/the-worlds-longest-running-refined-seed.html
asnwering partially why former eastern blog is faring so bad in terms of CVD. Also, causation would need correlation, and it does not exist...for diet-heart theory.
So delighted to have you back, Prof. Noakes. I allready checked the Foundation pages for the continuation...
Some editing points: Fig. 2 has lost it's bars. African tribes and Inuit, maybe Masai e.g. was meant?
Some questions and comments:
Malhotra from 1967 did not consider abundant pufa-6 as a source of harm? Presuming, even though sugar consumption was 7 fold in the north, both were small, below possible treshold for harm?
The French and Israel paradoxes seem to be opposite extremes in terms of sat / pufa percentages. They point to the same direction; diet-heart is not ok, sat fat is protective and pufa-6 not so. Said as a hypothesis, however the unique mechanistics of pufa-6 breakdown to 4-hne poison etc. is compelling (and they seem to accumulate with time!).
".. individual serum cholesterol and nutrient composition of diet cannot be found correlated without repeated surveys to reduce the effects of intra-individual variation. (77, p. 162)"
Dave Feldman has shown this by manipulating his own cholesterol values. His recipe for treating the "numbers": fast and 3-day sat fat feast. The liver sends/returns less energy in from of VLDL's. Like his visualization: VLDL is a ro-ro ferry, where triglycerides are the cargo and cholesterol the life rafts (maybe vitamins the stand-by passengers). The question arises, how transient are the cholesterol numbers in those several studies, and are they so transient, that the measurement can not be trusted as such...?
JR
Ancel Keys' Cholesterol Con, Part 5
2