The Framingham Heart Study: 1948-Present
Even before the National Heart Act had been signed into law, the U.S. Public Health Service had assigned a physician named Gilcin Meadors to draw up a proposal for a long-term epidemiological study that would kickstart U.S.-funded research into coronary heart disease (CHD) (Figure 1).

Figure 1: A letter to Meadors from Dr. Bert Boone of Temple University laid the groundwork for the Framingham Heart Study. Reproduced from reference 1 (p. 17).
Meadors’ proposal was “to study the expression of coronary heart disease in a ‘normal’ or unselected population and to determine the factors predisposing to the development of disease through clinical and laboratory exam and long term follow up.” His funding request for $94,350 included “funds to buy ashtrays for the smoking needs of the study’s staff members” (1, p. 3)!
The town of Framingham, Massachusetts, was chosen because of its close proximity to Boston and a cohort of Harvard cardiologists (including Paul Dudley White), all of whom quite naturally expected to enhance their global reputations by participating in a novel, well-funded study that was breaking new ground in heart disease research. For the scientists involved, there was only the promise of significant career advancements; there truly could be no downside — provided they stayed faithful to the hypothesis being tested. Like Ancel Keys a few years later, they were in the right place at precisely the right time.
However, there was one significant but covert danger in this comfortable arrangement of friends overseeing and participating in the work of their friends. It was that the biases of this small cohort of scientists, united in their desire to discover a fundamental truth which could, in turn, make each a medical icon, could be wrong — in which case, the error would be perpetuated and accepted as truth.
I’ve described the full findings of the Framingham Heart Study (FHS) in a previous column (2), but these findings are not those that are taught to — and more importantly remembered by — doctors, dietitians, and those in training to be doctors or dietitians.
Instead, the popular mythology is that the FHS confirmed both the diet-heart and lipid hypotheses because it found those who ingested more dietary fat had higher blood cholesterol concentrations, which in turn explained their higher rates of CHD. I delve deeper into this topic subsequently.
1950: Dr. John Gofman Formulates the Original Diet-Heart and Lipid Hypotheses
In a previous column (3), I described how already in 1950, John Gofman, MD, had formulated the diet-heart and lipid hypotheses (4) two years before Keys would commandeer the ideas as his own.
Gofman posed as a double challenge for Keys and his future disciples. First, Gofman was far more qualified than Keys to undertake research into the dietary and other factors causing heart disease. But perhaps more importantly, Gofman’s diet-heart hypothesis gave equal weight to dietary fats and dietary carbohydrates as the factors driving atherosclerosis and the development of CHD.
According to Gofman:
What is solidly established is that the Sf° 20-400 lipoprotein levels [i.e., blood triglyceride or VLDL concentrations] on the average, can be raised by increasing the dietary carbohydrate intake and can be lowered by decreasing it. … Furthermore, many individuals who are characterized habitually by some type of error in their metabolism that makes their Sf° 20-400 lipoproteins habitually extremely high will experience a marked reduction in the blood levels of these lipoproteins when the carbohydrate intake is lowered. (5, p. 123, my addition)
Gofman continues:
These same lipoproteins are essentially unaffected, in the average case, by changing from animal to vegetable fats. This information is extremely crucial, for in many individuals the risk of coronary heart disease comes primarily from the Sf° 20-400 lipoproteins [VLDL or triglycerides]. For such individuals, any attempt to lower heart attack risk by shifting from animal fat to vegetable fat in the diet would be illogical. There would be no reason whatever to expect any benefits since one would be changing the diet in a manner directed toward affecting the Sf° 0-20 [LDL] lipoproteins, which is not the problem at hand for these persons. For such individuals, the preventive efforts would have to be directed toward lowering the carbohydrate intake, which will, on the average reduce the Sf° 20-400 lipoprotein levels. With respect to the effect of carbohydrates on the Sf° 20-400 lipoproteins, it is a matter of the amount of carbohydrate that is eaten rather than the total number of calories ingested. For example, if one maintains individuals at exactly the same number of calories per day, so that they do not alter the weight in any way, but takes out some of the carbohydrates in their diet and replaces them by vegetable oil, one finds that the Sf° 20-400 lipoprotein levels will fall. Achievement of this result of lowering the Sf° 20-400 lipoproteins requires neither any alteration in caloric intake nor any alteration in body weight. (5, p. 124, my additions and emphasis)
Subsequently, in 1958 Gofman pointed out a key logical flaw that has since been ignored (6). He noted that a number of studies had found increasing the dietary intake of vegetable oils produced a fall in blood cholesterol concentrations, and this has been interpreted as beneficial. But the addition of vegetable oils also reduced total carbohydrate intake, and since carbohydrate increases the Sf° 20-400 lipoprotein levels, which contain approximately 13% of cholesterol by weight, the shift from a higher- to a lower-carbohydrate diet might be the real reason why increasing the intake of vegetable oils causes a reduction in blood cholesterol concentrations.
Thus, Gofman warned: “No consideration was given by them to the possibility that the lowering of cholesterol levels might have been the result of the simultaneous removal of a large amount of carbohydrate from the diet” (6, p. 277).
Gofman next describes the effects of a low-carbohydrate (100 g/day) diet in a 65-year-old male subject with a previous myocardial infarction (Figure 2).
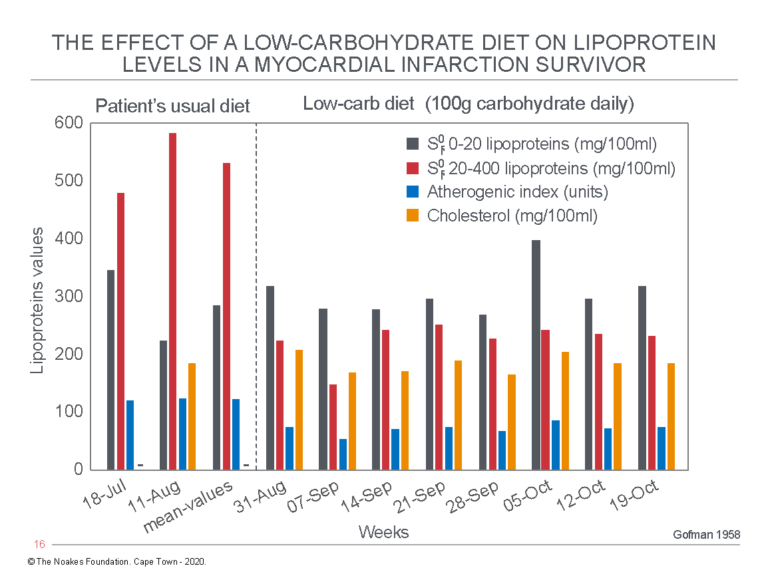
Figure 2: The effects of a low-carbohydrate diet in a myocardial infarction survivor. Note the low-carbohydrate diet produced a very large decrease in the Sf° 20-400 lipoprotein levels, now known as the VLDL-lipoproteins, which transport predominantly triglycerides. Total blood cholesterol concentration was unaffected by this dietary change. Despite this, the patient’s atherogenic index (AI) had fallen, placing him in a more favorable metabolic state according to Gofman’s understanding. Reproduced from data on Table V in reference 6, p. 279.
As Gofman wrote: “It can be seen from these data that a massive fall in the serum Sf° 20-400 lipoprotein levels occurs on the low-carbohydrate diet, without significant changes in the Sf° 0-20 lipoprotein levels. Accompanying this fall in lipoproteins is a highly marked and favourable reduction in the atherogenic index value” (6, p. 278-279).
Thus, the real originator of the diet-heart and lipid hypotheses stated that a low-carbohydrate, high-fat diet can be used in persons with established coronary atherosclerosis, presumably to reverse that disease.
He continued:
These same principles of carbohydrate restriction have been applied successfully in several types of extreme derangement of lipoprotein level control of the Sf° 20-400 lipoprotein class, namely, in xanthoma tuberosum, essential hyperlipidemia, and in diabetes mellitus … . For such a [post-myocardial infarction] patient, it is quite clear that management of the problem of coronary disease by dietary means involves the use of a low-carbohydrate diet, and not a low-fat, high-carbohydrate diet which is so often prescribed when attention is not paid to the lipoprotein findings. (6, p. 279-280, my emphasis)
The importance of this is that this evidence anticipated Peter Kuo’s “discovery” of carbohydrate-sensitive hyper(tri)glyceridemia (7) and its reversal with a low-carbohydrate diet by nine years (Figures 6 and 7 in reference 8).
In his conclusions Gofman wrote:
The increase in risk of future myocardial infarction associated with elevation of lipoproteins of the Sf° 20-400 lipoprotein classes provides the basis for a rational application of dietary measures in this disease … . Dietary carbohydrate intake is a prime factor controlling the serum level of the Sf° 20-100 and Sf° 100-400 lipoprotein classes. Restriction of dietary carbohydrates can provoke marked falls in the serum level of these lipoproteins … . The serum cholesterol measurement can be a dangerously misleading guide in evaluation of the effect of diet upon the serum lipids … . Rational management of patients with coronary heart disease or of individuals attempting to avoid coronary disease depends upon knowledge of the lipoprotein distribution in the individual patient. (6, p. 282-283)
Elsewhere Gofman wrote: “Neglect of [the carbohydrate factor] can lead to rather serious consequences, first in the failure to correct the diet in some individuals who are very sensitive to the carbohydrate action; and second, by allowing certain individuals sensitive to the carbohydrate action to take too much carbohydrate as a replacement for some of their animal fats” (9, p. 156-157).
In one of his last publications, a 1960 editorial, he again emphasized his concern about the carbohydrate factor:
Several investigators have shown that a low-fat high-carbohydrate diet produces opposite trends in the blood cholesterol and the blood lipid levels. The cholesterol level falls because the low fat diet depresses the level of the cholesterol rich Sf° 0-20 lipoproteins. The triglyceride level rises because the high carbohydrate intake elevates the level of the triglyceride-rich Sf° 20-400 lipoproteins. Both the triglyceride-bearing and cholesterol-bearing lipoproteins have been associated with the development of coronary disease. It therefore behoves the physician utilizing the dietary approach to understand the likelihood that a focus on the fat intake without an appreciation of the effect of carbohydrate intake will not lower all the blood lipids associated with the development of coronary heart disease. (10, p. 83)
1952: Kinsell Confirms Vegetable Oils Lower Blood Cholesterol Concentrations
In the early 1950s, Laurance W. Kinsell, MD, and his colleagues (11) were the first to publish evidence from human clinical experiments showing that the replacement of animal fats with vegetable fats produced large decreases in the blood concentrations of cholesterol and phospholipids.
A subsequent study by Mervyn Hardinge and Fredrick Stare (12) found blood cholesterol concentrations were lower in vegetarians who consumed no dairy products than in those who consumed eggs and milk or those who were not vegetarians.
Subsequently, J. Groen et al. (13) in the Netherlands found the blood cholesterol concentrations were independent of the total amount of fat ingested, since it was lower in vegetarians eating a higher-fat diet than in non-vegetarians eating animal fats but less total fat.
Later, a four-month long metabolic ward study published by Edward “Pete” Ahrens et al. (14) established that when fed vegetable or animal fats that provided the same total amount of dietary fat calories, the vegetable fats were associated with a 20% reduction in blood cholesterol concentrations. Thus arose the concept that saturated fats raise blood cholesterol concentrations more than vegetable fats.
As a result, the stage was being prepared for the replacement of animal fats with fats of vegetable origin in order to lower blood cholesterol concentrations. This was happening at the precise time Keys’ diet-heart hypothesis was beginning to gather intellectual support.
1953: A Study of U.S. Soldiers Killed in Action in the Korean War Reveals Many Have “Advanced” Coronary Artery Disease
Shortly after the outbreak of the Korean War, a group of U.S. pathologists was sent to the front lines with the gruesome task of performing autopsies on young U.S. soldiers killed in action. Their main objective was to determine the extent to which these young men showed evidence for coronary atherosclerosis and CHD (15).
The pathologists reported the results of 300 autopsies on soldiers in their early 20s. They reported 77.3% of the autopsied hearts showed gross evidence of coronary atherosclerosis. By this, I think they meant the atherosclerosis could be seen by the naked eye — that its presence was detectable even without the need for microscopic investigation.
Contrary to their initial reports, a more circumspect analysis showed that just 15.3% of the 900 arteries examined had arterial narrowings in excess of 40%, a level that would be considered important. Since each soldier had three main coronary arteries, this means 5-15% of the total sample of coronary arteries examined had significant coronary atherosclerosis — clearly not a “gross” finding.
While the authors restricted themselves to a discussion of their anatomical and pathological findings, resisting any hysterical conclusions, their findings raised some obvious questions: Without looking directly at their coronary arteries, how could those with advanced disease be identified? And if those at greatest risk could be identified, what does that knowledge tell us about the biological factors driving the development of this disease?
These would be the questions that would drive and that continues to drive the research program of the recently formed National Heart, Lung, and Blood Institute (NHLBI).
But was this study of U.S. soldiers meaningful? To draw conclusions from any scientific study, one needs a control group, but there was no control group in that experiment.
One potential control group, from a study published some years later, showed that the prevalence of atherosclerotic plaque in Japanese citizens was almost as high (65%) as in the U.S. Korean War casualties, even though the Japanese citizens had much lower rates of CHD and at the time were eating less fat and fewer animal products (16).
Dale Groom (16) also showed that whereas Haitians living in Haiti and African-Americans living in South Carolina had equivalent amounts of aortic atherosclerosis, African-Americans had more severe involvement of the coronary arteries. Groom wrote, “If the process is accelerated by the relatively high intake of cholesterol and fats in the American diet, one would expect the consequences in the aorta to parallel those in the coronary arterial tree. The reasons for the selective involvement of the coronaries are not known” (16, p. 57).
Similarly, the Japanese subjects had much higher mortality from cerebrovascular accidents (strokes) than the Americans, but they experienced fewer heart attacks (17). As Tavia Gordon notes, “We are faced with a serious problem. The present consensus seems to be that atherosclerosis is an important factor in both diseases [heart attack and stroke]. What mechanism would diminish [in the Japanese subjects] the effect of atherosclerosis on the coronary arteries while increasing its effect on the cerebrovascular system?” (17, p. 552, my additions). This paradox remains largely unanswered. Clearly, cholesterol-induced atherosclerosis is not the explanation to this paradox, particularly since in other populations, the extent of atherosclerosis in one artery parallels the extent of atherosclerosis in other arteries (18).
Another autopsy study found black South Africans showed significant aortic atherosclerosis (19), not dissimilar to that present in other South African racial groups who ate more meat and, at the time, had higher rates of CHD (20, 21). Similarly, Jack Strong, MD, and colleagues (22) reported black South Africans living in Durban, South Africa, had similar rates of fatty streaking of the aorta — the earliest manifestation of atherosclerosis — but more advanced stages of atherosclerosis, particularly fibrous plaques, were less common than in U.S. citizens. However, while rates of coronary atherosclerosis were clearly much less common, coronary atherosclerosis was not completely absent in black South Africans (23-25), Kenyans (26), or Northern Rhodesian mineworkers (27).
Recall that one of Keys’ very first studies (28) compared blood cholesterol concentrations in three different ethnic groups in my hometown, Cape Town (29). The authors of that study concluded: “The results of this study support the hypothesis that the dietary intake of fat influences the level of the serum cholesterol, particularly that in the Β-lipoproteins, and in turn may be one of the major factors influencing the pathogenesis of coronary heart-disease” (28, p. 1107, my emphasis).
Not so fast, Dr. Keys! To properly test your hypothesis, you should have checked whether the extent of coronary and aortic atherosclerosis matched differences in blood cholesterol concentrations in the different South African populations, because if it did not, your twin hypotheses are still-born.
That is the inherent problem with the use of surrogate measures (blood cholesterol concentrations) to predict a pathological process — in this case, coronary and aortic atherosclerosis.
Another study (30) found that whereas coronary atherosclerosis is more advanced in white (European) South Africans than in black South Africans, there was no difference in the extent of atherosclerosis in the cerebral arteries of the two ethnic groups. The authors felt there was an “absence of logical explanation” (30, p. 419) for this apparently anomalous finding. But the finding is only anomalous according to Keys’ hypothesis, which cannot explain why different arteries in the same individuals have different predispositions to develop atherosclerosis in response to the same blood cholesterol concentrations in all the arteries. Another study at that time confirmed that finding (31).
Jamaicans living in Jamaica had the same degree of atherosclerosis as a mixed population group in New Orleans; yet the incidence of myocardial infarction was much less in the Jamaicans (32). The authors concluded: “The lack of relationship between the incidence of atherosclerosis and ischemic heart disease does not suggest a common aetiology” (32, p. 444). In other words, heart attacks are caused by something other than or in addition to extensive coronary atherosclerosis — a point that has gone missing in the rush to declare “cholesterol” the sole cause of heart attacks.
The point is that epidemiological studies like those undertaken by Keys and his colleagues cannot conclude that the absence of clinical manifestations of CHD proves the absence of atherosclerosis as has been assumed.
The Masai tribesmen of Tanzania provide an interesting example of a population that eats a high-fat diet, but like other African peoples, the Sambura (33-35) and Anagamba (36) have low blood cholesterol concentrations (37, 38) and a near total absence of clinically apparent coronary heart disease (39, 40). Nevertheless, as George Mann et al. explained, they are not immune to atherosclerosis, and their “coronary arteries showed intimal thickening by atherosclerosis which equalled that of old U.S. men.” Mann et al. also noted, “The Masai vessels enlarge with age to more than compensate for this disease” (40, p. 26). Interestingly, blood cholesterol concentrations were not different in two African tribes, the one eating a diet providing 73% of energy from fat, the other just 9% (36).
Before the arrival of the modern industrial diet, the carnivorous Inuit also appeared to enjoy robust good health (41). As William Thomas, MD, explains:
These people live a life of great physical activity, a large number dying of violence, accident, starvation or freezing, before old age comes, exposed to the severest extremes of cold. They remain for hours and days in their kaiaks (sic), separated from icy water by only a membrane of sealskin. They frequently alternate between feast (when they eat to capacity) and famine. In view of these things, together with their extraordinary strength and endurance, the men often travelling twenty-four and thirty-six hours, continuously without rest or food, there can, I believe, be no other conclusion than that, under their conditions of life, an exclusive carnivorous diet does not predispose to renal or vascular disease. (41, p. 1559-1560).
The Inuit diet causes them to have predictably low blood triglyceride concentrations (42, 43) and slightly higher blood cholesterol concentrations than the African tribesmen studied by A. G. Shaper et al. (42). Some Inuit demonstrated evidence of mild atherosclerosis (44, 45), but “the severity was usually not enough to produce clinically recognizable symptoms and signs” (42, p. 743). As a result, CHD is uncommon (46), perhaps because the diet is high in omega-3 polyunsaturated fatty acids from marine mammals (47, 48), which may be associated with longer bleeding times and reduced platelet aggregability (49).
Another study that is ignored is the 1968 International Atherosclerosis Project, which collected information on 12 populations in the U.S., South and Central America, Africa, Europe, and the Philippines (50, 51). The study found that between the ages of 25 and 34, similar degrees of early atherosclerosis (raised arterial lesions) were present in virtually all the different populations across the world, regardless of whether their diets were predominantly meat- or plant-based, and whether or not the populations had high or low rates of CHD (51). The authors wondered whether arterial stenosis — which produces the clinical symptoms we recognize as CHD (i.e., heart attacks, sudden death, angina pectoris) — “may result from thrombosis rather than the growth of atherosclerotic plaques, and [whether] stenosis may result from a process concurrent with but pathogenically independent of earlier stages of atherosclerosis” (51, p. 39). In other words, thrombosis contributes to atherosclerosis and clinical CHD but is not included in Keys’ simplistic hypotheses about how CHD develops.
The 1968 International Atherosclerosis Project also found that although there was a correlation between blood cholesterol concentrations and the extent of atherosclerosis, no relationship was found between the amount of animal fat consumed and either blood cholesterol concentrations or the extent of atherosclerosis (50, 51). Thus, the study authors concluded: “The estimated percentage of total dietary fat consisting of animal fat does not appear to be associated with severity of atherosclerosis” (51, p. 499). They added: “In most populations sources of fat and of carbohydrate are not the primary factors in determining severity of atherosclerotic lesions” (50, p. 627). Essentially, the same findings would be confirmed by the Framingham Heart Study and the Seven Countries Study.
Finally, there is the study by R. P. Malhotra and N. S. Pathania (52), which addressed the then-popular belief that CHD is rare among Indian populations who are predominantly vegetarian, eating animal products only rarely. Malhotra and Pathania concluded CHD was already a growing problem in the Indian community in 1958 and explained, “We find no reason to believe that vegetarians are less predisposed to coronary disease” (52, p. 530).
All these findings are difficult to reconcile with the diet-heart and lipid hypotheses.
But unlike William Enos and colleagues’ study of the U.S. Korean war casualties (15), which has achieved iconic status and is known to many, all these inconvenient findings are simply ignored.
The ignored studies have one thing in common: They imply atherosclerosis is driven by factors other than cholesterol and a high-fat diet and indicate the extent to which coronary atherosclerosis is present in anyone does not necessarily predict the likelihood that he or she will develop a clinical manifestation of that disease, specifically by experiencing a heart attack or stroke.
In other words, atherosclerosis, heart attacks, and strokes may not all be caused by identical mechanisms. And what if cholesterol is not the most important driver of any of them?
1953: Keys Happens Upon a World Health Organization Report Showing a Relationship Between National Dietary Fat Intakes and Rates of Heart Disease
In previous CrossFit columns (2, 3, 54-58), I’ve dealt in some detail with Keys’ central role in the popularization of the diet-heart and lipid hypotheses. I have also listed his many scientific misdemeanors.
The most salient points are that Keys promoted evidence from associational epidemiological studies as if such studies could prove causation — even though he knew full well this is not the case. When his first RCT, the Minnesota Coronary Experiment (MCE), failed to show an expected benefit as a result of removing saturated fat from the diet, Keys and his colleagues simply ensured the data went missing.
Keys chose this route because he lacked the scientific training to add any meaningful new knowledge. Instead, he stole Gofman’s ideas and proceeded to write Gofman out of history. The result was that Gofman — whose pioneering work showed he had a more complete understanding of the biological effects of dietary carbohydrates and fats on the blood lipid fractions he believed were important in the development of coronary atherosclerosis — simply lost interest in the field. He opted out and moved on to study other phenomena.
As a result, Gofman’s understanding that high-carbohydrate diets could be harmful (5, 6, 10) because they increased blood triglyceride concentrations (VLDL; Sf° 20-100 and Sf° 100-400) was largely forgotten until his contribution was again recognized by the investigative writing of Gary Taubes (9).
The problem was that because dietary fats, especially saturated fats, had been demonized by Keys and his acolytes, replacing them with carbohydrates and vegetable oils would have to be immunized from scrutiny. In effect, the carbs and vegetable oils would need to be “health washed.” But this could not occur if the medical and scientific communities had any suspicion that carbs might be linked to CHD, as Gofman was suggesting, or that vegetable oils might be linked as well, as Enig and Kummerow had argued (58).
The only solution was to ignore Gofman’s findings, which, as I will show, duly happened at the National Consensus Development Conference in 1984 and with the subsequent launch of the National Cholesterol Education Program in 1986 (59). Both excluded any mention of the work of Gofman. They also did not mention Donald Fredrickson, Robert Levy, and Robert Lees, whose pioneering studies had confirmed, perhaps even more convincingly than those of Gofman, that carbohydrates drive the (carbohydrate-sensitive) hypertriglyceridemia present in perhaps the majority of persons with CHD.
As a result, Keys’ simple model based on one simple WHO diagram (Figure 2 in reference 29) that by itself proved nothing would become the globally accepted explanation for how diet can cause coronary atherosclerosis.
It became all about dietary saturated fat and cholesterol in the blood, at the exclusion of all other possible factors.
Eisenhower Suffers a Heart Attack and Is Prescribed Untested “Heart-Healthy” Low-Fat Diet
I’ve described the historical importance of President Eisenhower’s heart attack in an earlier column (60). The main point is that one of the most important individuals in the world was placed on an experimental low-fat, supposedly “heart-healthy” diet before there was any evidence that the diet was beneficial and not harmful. The sole assurance the president had was his complete faith in the nation’s most revered cardiologist, Paul Dudley White, and, in turn, White’s unwavering trust in Keys and his untested hypothesis.
In the end, the president’s faith proved to be misplaced, for the dietary advice he received was clearly harmful. Unfortunately, Eisenhower, in keeping with his military background, was the perfectly compliant heart patient. He did exactly as he was told. On this experimental diet he developed Type 2 diabetes mellitus (T2DM); his coronary atherosclerosis progressed remorselessly; and he died of intractable heart failure.
If there was ever a single anecdote disproving Keys’ hypothesis, this was it.
But this was not sufficient to cause either Keys or White to pause and question their own beliefs. Instead, under their influence, the president became a strong advocate of the AHA. As Nina Teicholz describes, during the remaining years of his presidency, Eisenhower presented the AHA’s annual “Heart of the Year Award” from the Oval Office in the White House. He also attended AHA board meetings and served as the AHA’s Honorary Chairman of the Future (61, p. 48-49). Other members of Eisenhower’s cabinet also served on the AHA board.
Thus the AHA itself — captured by the food industry and especially companies manufacturing foods that had never been eaten in the 4-6 million years of the emergence of Homo sapiens from our hominid ancestors — had in turn captured significant influence in the U.S. government.
1955: Seminar of the World Health Organization (WHO) Study Group on Atherosclerosis and Ischemic Heart Disease
As I described previously (29), Keys’ first presentation of his soon-to-become-iconic 1953 paper (62) was at the WHO Study Group on Atherosclerosis and Ischemic Heart Disease in 1955. There, his work was torn apart by Jacob Yerushalmy and Herman Hilleboe, who two years later published the criticisms they had presented at the meeting (63).
The event was pivotal for Keys. It seems it was the only time anyone had the courage to stand up to him. His response was predictable. As Teicholz explains, Keys “got up from being knocked around and said, ‘I’ll show these guys’… and he designed the Seven Countries study” (61, p. 36).
This is certainly not the best way to begin a research career, the goal of which, outwardly at least, should have been to discover the optimum human diet. Clearly Keys’ goal had become something quite different.
His goal instead became to prove that he was right and Yerushalmy and Hilleboe (who were clearly his intellectual superiors) were wrong. That attitude conflicts absolutely with the methodology scientists are meant to adopt, which is to prove that their most deeply held biases are incorrect — the concept of the null hypothesis.
But, as history has proved, for Keys this was never going to happen. Instead, he was preparing to embark on a sustained, life-long campaign to secure his legacy as the most correct and influential nutrition scientist in history. To achieve that, he was willing to do whatever it took, so he became perhaps the world’s single greatest example of a scientist intoxicated with confirmation bias (9, 61). This means he was only capable of seeing the evidence that supported his own belief systems. All contrary evidence was to remain unseen.
1956: U.S. Public Health Service Funds Keys’ Seven Countries Study
Keys’ Seven Countries Study (SCS) (54) went on to achieve iconic status as the first multi-country epidemiological study of the relationship of diet to heart disease. It achieved this iconic status even though it had a number of fatal flaws that prevented it from ever coming to any definitive conclusions.
The results were first released in 1970, as we discussed earlier (54) and will discuss in greater detail subsequently.
1957: AHA Nutrition Committee Finds No Strong Evidence to Support Keys’ Diet-Heart Hypothesis
After Keys’ presentation at the WHO meeting and due to the growing influence of food companies like Procter and Gamble, it was predictable that the AHA would need to make a public statement on any potential role of diet in the causation of the heart disease epidemic. This was especially true following President Eisenhower’s heart attack and the national and international interest that event had triggered. Two official publications in 1957 by eminent U.S. physicians reviewed the state of knowledge at that time.
An official report (64) to the AHA from a committee chaired by Irvine H. Page, MD, concluded:
In the opinion of the authors of this review, there is not enough evidence available to permit a rigid stance on what the relationship is between nutrition, particularly the fat content of the diet, and atherosclerosis and coronary heart disease. We are certain of one thing: the evidence now in existence justifies the most thorough investigation. This should be done soon, thoroughly, and uncompromisingly. (64, p. 164)
The authors argued, “The proposition that the character of the American diet has so changed during the past 50 years as to increase the incidence of coronary vascular disease cannot be supported” (p. 171). Citing different studies, they provided evidence that different groups of Americans between 1891 and 1953 were eating diets with up to 46% of calories derived from fat. For example, a study of lean and obese adult women in the U.S. found that fat provided between 36-46% of their daily calories in 1953 (65).
But the reality is that modern Americans began to eat substantially less meat in the 100 years immediately before CHD rates began to increase (Figure 2) (66, 67) and especially in the 1930s after the Great Depression (66, 68).
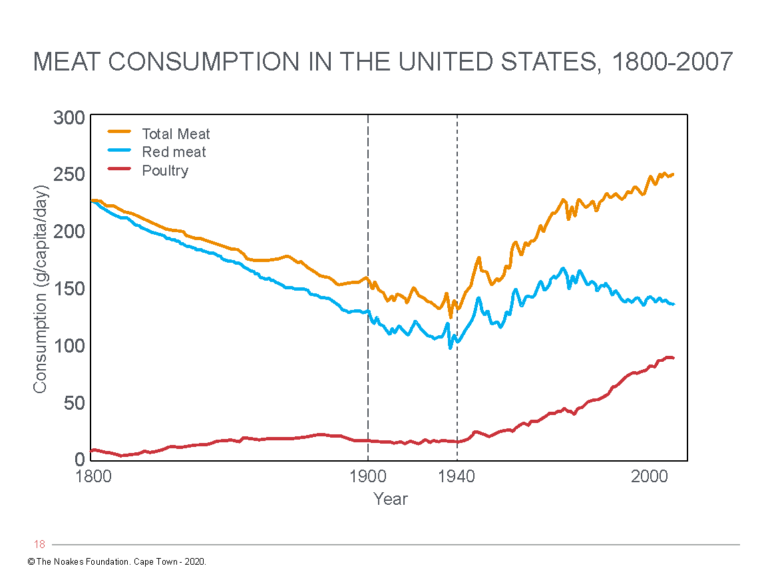
Figure 3: Trends in daily consumption of total meat, red meat, and poultry. Notice in the period when CHD rates began to significantly increase (1900-1940), red meat consumption was falling and was perhaps 50% lower than amounts eaten in the 1800s in the U.S. After 1940, red meat consumption again increased while CHD rates were starting to fall (after the 1950s). Poultry consumption has increased dramatically since the end of World War II (1945). The data contradict Keys’ central assertion that the increased consumption of saturated fat in meat is the key driver of the CHD epidemic of the 19th Century. Reproduced from reference 61, p. 115.
As Roger Horowitz explains, “Meat consumption [in the U.S.] dropped to unusually low levels in the Great Depression … . Consumption rebounded in the 1940s, but it remained well below levels seen in the early twentieth century and was sharply distinguished by income” (66, p. 15). He continues: “Meat consumption began to climb dramatically in the 1950s after the end of the Korean War’s rationing program. By 1965 it had reached the highest levels in [recorded] American history ” (66, p. 15).
Importantly, meat intake has always been least in the lowest socioeconomic classes, precisely among those who suffer the greatest burden of chronic diseases including CHD (69).
Teicholz concludes that in the 1800s, Americans were each eating between about 150-200 pounds of red meat annually, compared to about 100 pounds in the 1940s (Figure 3), immediately before the CHD epidemic was first recognized (61, p. 115). Today, red meat consumption is not greatly different from the 1940s, whereas total meat consumption has returned to amounts last eaten in the 1800s.
However, the real increase in meat consumption in the U.S. after 1940 comes almost entirely from an increase in poultry consumption (Figure 3).
Thus, the conventionally described increase in CHD rates in the U.S. from 1910 to 1950 was associated with decreased meat (and hence saturated fat intake from meat), whereas declining rates after the 1960s occurred subsequent to a ~50% increase in meat consumption, especially in those in the lowest socioeconomic groups. Most of this increase resulted from an increased consumption of white meat, especially poultry (Figure 3).
Teicholz points out that historians Waverly Root and Richard de Rochemont wrote, “It would be incorrect to describe Americans as great eaters of either [fruits or vegetables]” (61, p. 117). Teicholz concludes: “So by these accounts, for the first two hundred and fifty years of American history, the entire nation would have earned a failing grade according to our modern mainstream medical advice” (61, p. 117). In addition, she claims that if increased meat consumption causes heart disease, “there were some ten million Americans of prime age for having a heart attack at the turn of the twentieth century, but heart attacks appeared not to have been a common problem” (p. 118).
The other truly critical point that had to be hidden by the Keysian acolytes is this: “From the outset, the validity of the diet-heart hypothesis, linking the conventionally described rising CHD incidence after 1910 to an increased saturated fat intake from meat especially, is not supported by the evidence, at least in the USA … . Instead, it would seem to be disproven by this information that is conveniently ignored by advocates of the diet-heart hypothesis” (61, p. 23).
Although she does not refer to it, Teicholz’s conclusion is supported by the conclusions of Dr. Mary Enig, who already in 1977 showed that as a percentage of total fat intake, Americans’ animal fat intake — the main source of saturated fat intake — has been in decline since 1909 (Figure 2 in reference 70). Conversely, total animal fat intake did not change materially between 1909 and 1957 (Figure 1 in 70), though there was some decline thereafter. What has changed dramatically is the contribution of polyunsaturated fat from vegetable oils (Figures 1 and 2 in reference 70), and there has been a marked change in the dietary polyunsaturated fat-to-saturated fat ratio (Figure 3 in reference 70).
Page and his committee were absolutely correct to argue that “a wide variety of other factors, dietary and nondietary, may be of equal or greater importance, and … that infarction, the real nub of the problem, is not generally produced experimentally, despite the extensive and severe atherosclerosis that has been produced [in animal models]” (64, p. 174).
Their more specific point was that there is a further step before atherosclerosis of the coronary or cerebral arteries initiates a heart attack or stroke, and that is rupture of the atherosclerotic plaque with the formation of an obstructive blood clot. So they asked: “Is there compelling evidence that, if we treat the hypercholesterolemia by dietary means, we are doing anything to lessen the chance of myocardial infarction?” (64, p. 174). In other words, does lowering the blood cholesterol concentration reduce the risk of plaque rupture (as opposed to preventing further growth of the obstructing plaque)?
In their conclusions they wrote:
Thus, the evidence at present does not convey any specific implications for drastic dietary changes, specifically in the quantity or type of fat in the diet of the general population, on the premise that such changes will definitely lessen the incidence of coronary or cerebral artery disease. On the other hand, the fact that obesity is a nutritional failure, that it is caused by consuming more energy than one expends, that dietary fats are the most concentrated source of energy, providing some 40 to 45 per cent of the daily caloric intake, suggests that many should consume less calories. For most, this will mean eating less fat. (p. 175)
They also wrote, “Perhaps the best that can be said is that there is an association that has statistical value, but that is not an obligatory association either in small groups or, and much less so, in an individual” (p. 174).
“The key points of nutritional common sense for better health generally and most likely in regard to atherosclerosis specifically, consist of a balanced, varied diet that adjusts total calories to reach or maintain a desirable weight,” they argued. “Such a diet should provide more protein from lean meat, fish, poultry, and animal products, cereal and grain products, and a reasonable selection of fruits and vegetables. The fat content should be sufficient only to meet caloric and essential fatty acid demands” (p. 175).
Unfortunately, Page and his co-authors introduced an error of logic: They claimed that because dietary fat contains more calories than does carbohydrate, eating too much fat is the cause of the growing obesity epidemic. Thus began the error of the “balanced, low-fat, heart-healthy, prudent diet, in moderation” even before there was any evidence that eating less fat would prevent the development of obesity. Today, we know this advice is wrong, because removing fat from the diet increases hunger and the resulting increased caloric consumption is one of the key drivers of the modern obesity and diabetes pandemics.
In retrospect, the authors made one other serious error in their review; an error that would cause long-term damage to the health of the world. They ignored that, at the time, there were two competing theories for what causes heart disease: Keys’ diet-heart and lipid hypotheses stood in opposition to Gofman (5,6,10), Albrink and Mann (71), and later Ahrens (72), Kuo (7), and Fredrickson’s (73) evidence that carbohydrate-sensitive hyper(tri)glyceridemia (CSHT) and T2DM were at least as likely to be causative. As a result, their report (64) unfortunately ignores any potential role of carbohydrates in causing obesity, T2DM, or atherosclerosis.
The AHA and these authors failed because they should have proposed in 1957 that there were two competing theories to explain why coronary atherosclerosis develops. What subsequent events have taught is that by funding research of only one of these two competing options, the inevitable happened: That theory became the one that dominated, simply because it carried all the power, influence, egos, and money.
By studying only one of the two possibilities, the AHA and NHLBI established an institutional bureaucracy that favored the confirmation that the hypothesis in which they had invested so heavily had to be the correct one. Since all the money was supporting only one hypothesis, these institutions naturally attracted and rewarded only those scientists — like Keys, Stamler, Steinberg, Kannel, Castelli, and others — who believed that hypothesis was correct. A scientist who suggested otherwise might have to endure a fatal blow to their career, as was the case for Yudkin and Mann (29).
As Teicholz has argued, research on heart disease became a matter not of science but of the institutionalization of a false and disproven theory:
Thus, the normal defenses of modern science had been flattened by a perfect storm of forces gathered in postwar America. In its impressionable infancy and compelled by an urgent drive to cure heart disease, nutrition science had bowed to charismatic leaders. A hypothesis had taken center stage; money poured in to test it, and the nutrition community embraced the idea. Soon there was very little room for debate. The United States had embarked upon a giant nutritional experiment to cut out meat, dairy, and dietary fat altogether, shifting calorie-consumption over to grains, fruits, and vegetables. Saturated animal fats would be replaced by polyunsaturated vegetable oils. It was a new, untested diet — just an idea, presented to Americans as the truth. Many years later, science started to show that this diet was not very healthy after all, but it was too late by then, because it had been national policy for decades already. (52, p. 102)
1957: Ahrens and Colleagues Publish a Review of Dietary Factors That Influence Blood Cholesterol Concentrations
Ahrens, one of the four co-discoverers of carbohydrate-sensitive hyper(tri)glyceridemia (CSHT) (72), responded to the AHA document and included a review of his team’s work on dietary factors influencing the blood cholesterol concentrations (74).
He and his colleagues began with the disclaimer: “There is a serious lack of direct evidence that atherosclerosis is due to alterations in fat metabolism” (74, p. 1905).
Included in the article is a figure (Figure 4), the logic of which continues to be largely ignored by those promoting Keys’ hypotheses.
The figure makes the simple point that there is a sequence of events that occur between “bad” dietary fat and coronary disease, and that at each of these three steps, multiple factors are likely to be influencing the final outcome. Such factors represent confounders, of the kind that epidemiological studies like the Seven Countries Study are highly unlikely ever to uncover.

Figure 4: The pathway by which “bad” dietary fat leads to coronary disease according to Keys’ twin hypotheses. The arrows indicate confounding factors that might be acting at points 2-4 to produce the final outcome of CHD. Particularly important but often ignored are the factors that convert atherosclerosis to a clinical endpoint such as a heart attack, stroke, or death. To go from point 3 to point 4 requires rupture of the atherosclerotic plaque with development of blood clotting in the affected coronary artery — historically referred to as coronary thrombosis. Factors that produce this outcome are not included as any possible components of Keys’ hypotheses. Reproduced from reference 74, p. 1906.
As Ahrens and colleagues note:
To us, the most serious weakness of this type of evidence lies in our inability at the present time to quantitate the extent of the atheromatous process during life. Not only does this weak link prevent an evaluation of the extent of the disease process in relation to abnormal serum lipids and antecedent dietary malpractice, but it also prevents the direct estimation of the effectiveness of any regimen that might ameliorate the process. It is regrettable that the good and bad features of any environmental factor cannot be evaluated except in terms of the complications of the atherosclerotic process. (74, p. 1906)
Their argument is that diet is but one factor that could be driving the progression from point 1 to point 4, and since the only measurable hard endpoint (death, heart attack, stroke) is point 4 in that figure, it becomes extremely difficult to be certain that the hard endpoint can be ascribed solely to point 1 – eating “bad” dietary fat. What about all the other potential confounding factors acting on points 2-4?
The authors therefore warned:
It is entirely possible that the scheme shown in figure (3) will be proved correct when further facts have been gathered through new approaches to the problem. However, when unproved hypotheses are enthusiastically proclaimed as facts, it is timely to reflect on the possibility that other explanations can be given for the phenomena observed. (74, p. 1906)
By supporting only one of two competing hypotheses, the AHA and NHI had effectively shut the door on research exploring those other possible explanations.
The article included perhaps the first detailed review of the effects of different dietary fats on blood cholesterol and other lipid measurements. Their summary findings were:
The highest levels of (blood) cholesterol are found when butter and coconut oil are fed as sole dietary fats, intermediate levels are produced by palm oil, lard, cocoa butter, and olive oil; the lowest levels following feeding of peanut, cottonseed, corn and safflower oils. A linear correlation was found between the saturation of the dietary fat and the levels of cholesterol and phospholipids in the serum. (74, p. 1907)
These findings became the basis for Keys’ prescription of vegetable oils for the prevention of CHD. This explains why Keys’ first trial, the Minnesota Coronary Experiment (MCE), replaced (some) dietary saturated fats with polyunsaturated vegetable oils rich in linoleic acid.
But even then, Ahrens et al. had noted:
Since in some patients a diet low in fat produces high levels of serum triglycerides [due, as Gofman would point out, to the higher carbohydrate intake], we are tempted to ask whether the lower-density lipoproteins are less “atherogenic” than the higher density lipoproteins rich in cholesterol and phospholipids. We know of no solid evidence on this point, and until the question is further explored we question the wisdom of prescribing low-fat diets for the general population. (p. 1909)
In essence, the authors were reiterating what Gofman (5, 6, 10) and later Fredrickson (73) had warned against: Don’t ignore the potential role of carbohydrates in causing atherosclerosis and CHD. Unfortunately, such warnings were simply written out of the record as the move to incriminate dietary saturated fat and elevated blood cholesterol concentrations intensified.
Ahrens et al. concluded:
In view of the fact that it has not been demonstrated in man that lowered levels of serum lipids will alter his susceptibility to atherosclerosis and in view of the many questions that remain unanswered in this year 1957, it seems inadvisable at this time to recommend that the general public eat more or less fat or avoid fat of any kind. We believe that much more basic information must be gathered before widespread changes in dietary habits are adopted … .
The hypothesis that dietary fats affect atherogenesis, however plausible and appealing, remains unproven. Serum lipids can be altered by dietary means, and experimental data lead to the presumptive conclusion that unsaturated fats in the diet cause depressions in levels of cholesterol and phospholipids. Serum triglycerides, on the other hand, can be made to rise on low fat diets and fall when fat intakes are high. Thus, serum lipids are affected by both quantity and quality of ingested fat. Radical changes in dietary habits are not recommended to the general public at this time. However, patients with existent or threatening atherosclerosis may be justifiably advised to eat high portions of unsaturated fats. (p. 1911)
1957-1972: The Anti-Coronary Club Program
While he was Director of the Bureau of Human Nutrition at the New York City Department of Health, Norman Jolliffe, MD, first read about the low-fat diet Keys had prescribed for President Eisenhower (60). Intrigued, he decided to initiate what might have been the first clinical trial of a low-fat diet enriched with polyunsaturated fats in the management of patients who had suffered a heart attack. Later, he would also use his experience to write a popular book called Reduce and Stay Reduced on the Prudent Diet (75).
Jolliffe’s trial became known as the Diet and Coronary Heart Disease Study Project, or more popularly as the Anti-Coronary Club Program in New York City. The study began in 1957 and ran until 1972 (76-83). During that period, 1,113 patients were placed on an experimental diet — the original Prudent Diet, which limited beef, mutton, and pork consumption to four meals a week. The remaining meals comprised poultry and fish, with the latter to be eaten at least four times a week. Butter and hydrogenated shortening were replaced by PUFA-enriched margarines, and one ounce of vegetable oil was ingested daily. The control group of 467 continued to eat their usual diets.
Unfortunately, in 1961, while the trial was still ongoing, Jolliffe died suddenly of a heart attack.
While the clinicians who continued the trial after Jolliffe’s death claimed his Prudent Diet reduced the number of coronary events, hidden in their data was one very inconvenient fact: Whereas there were 18 deaths from non-coronary causes in the group eating the Prudent Diet, there were only six such deaths in the control group that continued to enjoy their dietary saturated fats (81, p. 601).
Clearly the Prudent Diet had not helped the Anti-Coronary Club members live any longer. It was not an auspicious start to the testing of the diet-heart hypothesis.
Jolliffe left one other contribution that also has been ignored. In an article published in 1959 (77), two years before his untimely death, he included a figure that addressed some of the concerns raised by Ahrens and colleagues (Figure 5).
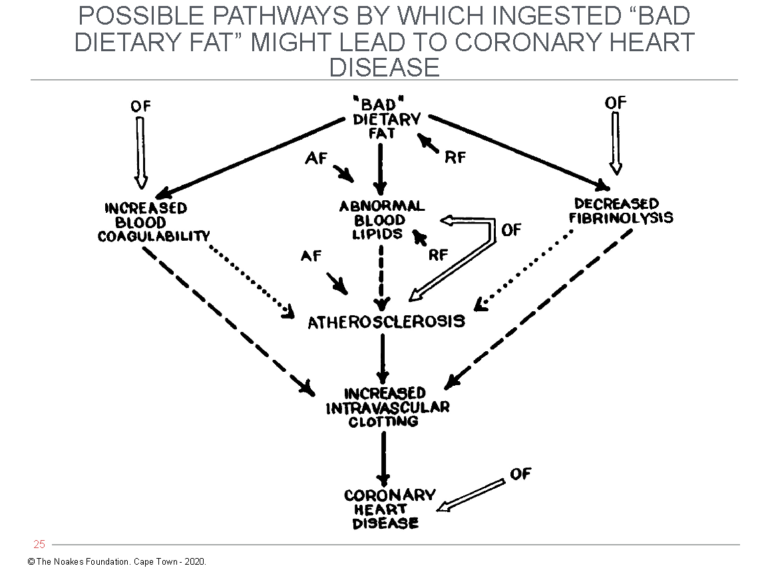
Figure 5: Jolliffe’s expansion of Keys’ simplistic diet-heart hypothesis. As Ahrens and colleagues had argued in 1957, a number of factors might be involved in turning “bad” dietary fat into CHD (Figure 4). Importantly, Jolliffe included the development of “increased intravascular clotting” as the final step in the pathway, which is logical since coronary atherosclerosis alone cannot cause the clinical manifestations of CHD. This continues to be ignored by those dedicated to the dogma of the diet-heart hypothesis. (AF = Accentuating Factors – e.g., genetics, hormones, diabetes, kidney disease, maleness. RF = Retarding Factors – e.g., genetics, hormones, femaleness; OF = Other Factors — i.e., those outside the scheme and unrelated to dietary fats.) Reproduced from Figure 4 in reference 77, p. 124.
Figure 5 shows that theoretical accentuating factors (AF) and retarding factors (RF) may act at different points along the pathway from “bad” dietary fat to CHD. Jolliffe also added the key component of increased intravascular clotting, which is completely absent from Keys’ simplistic model.
What is remarkable is how this eminently reasonable model is never promoted as a more probable explanation than the reductionistic “cholesterol” model for how diet, or in fact many other accentuating or retarding factors, can lead to CHD.
How the entire academic world was brainwashed into adopting a reductionistic model in which just one causative factor could be considered of overwhelming importance is the subject we tackle in the following columns.