Table 1 in the previous column (1) lists the sequence of events that, I argue, were critical in directing the global acceptance of Ancel Keys’ unproven hypotheses, the diet-heart and lipid hypotheses.
In this column, we investigate those events that happened between 1910 and 1948.
1910: Future Nobel Laureate Adolf Windaus Detects Presence of Cholesterol in Arterial Atherosclerotic Plaques
In 1910, as part of his pioneering studies of the role of cholesterol in human metabolism, German chemist Adolf Otto Reinhold Windaus discovered atheromatous arterial lesions (arterial plaques) contain six times as much free cholesterol and 20 times as much esterified cholesterol as do healthy arteries (2). Windaus would also later describe the pathways by which cholesterol is converted to vitamin D. For his work, he was awarded the Nobel Prize in chemistry in 1928.
The assumption at the time was, predictably, that the cholesterol in arterial plaques must arise from the cholesterol circulating in the bloodstream. In time, this finding gave rise to Gofman/Keys’ lipid hypothesis, which holds that elevated blood cholesterol concentrations (caused by eating a high-fat diet especially rich in “artery-clogging” saturated fats) drive cholesterol across the arterial lining, a single layer of cells known as the endothelium, and into the subendothelial space, causing the initiation of fatty streaks (Figure 1). These then progress to the development of more advanced atherosclerosis, termed arterial plaques (Figure 2), which among other complications can cause heart attacks and strokes.
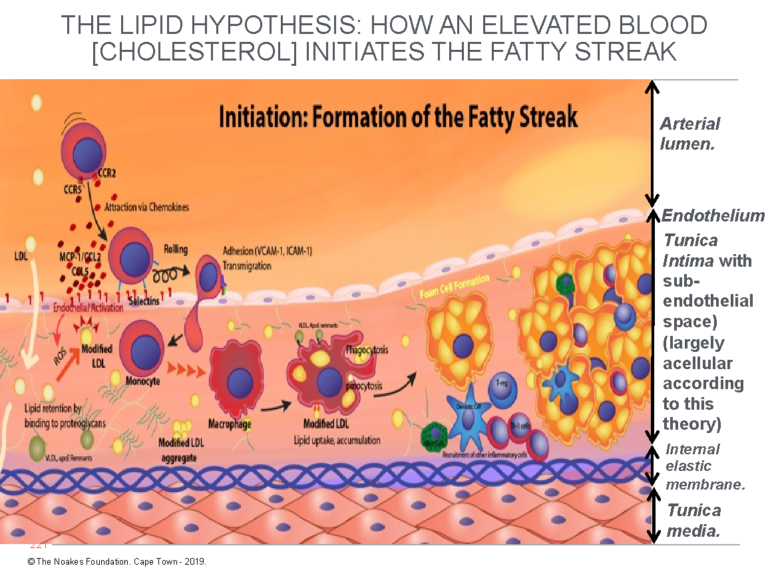
Figure 1: This diagram explains the currently accepted theory of how endothelial damage, largely of unknown cause, allows LDL-cholesterol to cross the endothelium and enter a postulated and hypothetical acellular space, the subendothelial space. There, the LDL-cholesterol is taken up by macrophages, causing the development of the earliest form of atherosclerosis, known as the fatty streak. Note that for the atherosclerotic process to happen in this way, the tunica intima must be devoid of all cells other than the single layer of endothelial cells that coat its upper surface, separating it from the blood contained in the lumen of the artery. According to this model, the subendothelial space is essentially a wide-open vacant space waiting expectantly to accommodate these (complex) processes that produce atherosclerosis. Reproduced with additions from reference 4, p. 3.
The finer details in Figure 1 are not critical to the argument. What is important is the way in which the different cellular structures are depicted.
Here, the crucial point is that, according to the currently popular explanation of atherosclerosis (4), until the endothelium is damaged (by currently unknown biological events), allowing the unrestrained entry of LDL-cholesterol, the tunica intima is depicted as a single, thin layer of endothelial cells sitting on top of an acellular space (devoid of cells). This is an important consideration, since the presence of any cells in the subendothelial space must impede the entry of LDL-cholesterol directly from the bloodstream and will hinder the ability of the macrophages to detect and consume the cholesterol, as depicted in Figure 1.
Figure 2 depicts how this model explains the progression of the fatty streak to full-blown atherosclerotic plaque. Note again that the subendothelial space is devoid of cells before the hypothetical endothelial damage allows the free entry of LDL-cholesterol into this conveniently located anatomical space.
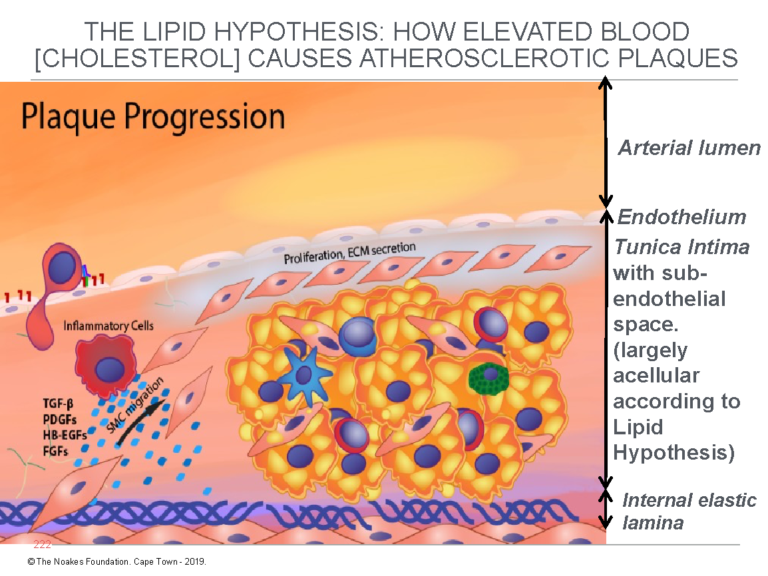
Figure 2: This figure shows how the fatty streak (Figure 1) progresses to the atherosclerotic plaque according to the lipid hypothesis. For the lipid hypothesis to be true, until after the initial “injury” to the endothelium has allowed the entrance of blood-derived LDL-cholesterol, the tunica intima must, as shown in this figure and in Figure 1, contain no cells other than the thin layer of endothelial cells on its upper surface. Notice that in this figure, smooth muscle cells (SMC) migrate from the tunica media into the tunica intima to further progress the development of the atherosclerotic plaque. Reproduced with additions from reference 4, p. 8.
In 1910, neither Windaus nor anyone else was aware that cholesterol cannot simply pass through healthy arterial walls, however hard it may be “shoved” (3). Currently, the most popular theory for atherosclerosis is that shown in Figures 1 and 2. This theory holds that the endothelial cells lining the lumen of the artery wall must first be damaged before the passing of cholesterol through the wall can happen. This is termed “endothelial cell dysfunction” (4), but the immediate cause of “endothelial cell dysfunction,” if this is indeed the mechanism, remains shrouded in secrecy even today, 110 years after Windaus’ discovery.
This theory also predicts that cholesterol enters damaged arteries down a concentration gradient, so the degree of a person’s arterial disease can be predicted quite simply as their average blood cholesterol concentration multiplied by the number of years the blood cholesterol concentration has been “elevated” (3, 5).
Also, still unknown then was that atherosclerosis is a patchy disease that selectively targets only specific areas of different arteries. This is exemplified by what happens in the coronary arteries supplying blood to the heart muscle (6).
It also was then unknown that in some populations, there may be advanced atherosclerosis in the cerebral (brain) arteries with minimal involvement of the coronary (heart) arteries (7, 8). In such cases, a person is at greater risk of stroke than heart attack. In other cases, as is more prevalent in the U.S., the opposite applies.
So, even today, devotees of the lipid hypothesis seldom ponder the inconvenient question: If the same (elevated) blood cholesterol concentration bathes the 100 or more miles of blood vessels in the adult human, why is it that only small segments of the arteries ever become diseased? How is it possible that there is some magic borderline dividing those cells that will become atherosclerotic from those that are apparently immune to the (supposedly) direct effects of elevated blood cholesterol concentrations? And why are veins so seldom affected except in cases where they are used to bypass diseased coronary arteries?
The greatest challenge to this traditional explanation has been presented by Professor Vladimir Subbotin (9-11). The challenge hinges on two facts that are irrefutably true, and either of which by itself irretrievably undermines any proposed theory of how LDL-cholesterol from the blood crosses the lining of the arteries — i.e., the endothelium — and collects in the subendothelial space in the tunica intima, initiating the process of atherosclerosis as depicted in Figures 1 and 2.
The first point made by Subbotin as early as 2012 (9) and repeatedly since (10, 11) is that the tunica intima, including the so-called subendothelial space, is not an empty space without cells and filled only with structural proteins. This is the way it is depicted in Figures 1 and 2. Subbotin argues the only reason why the diagram is drawn that way is because Keys’ lipid hypothesis demands it to be so. Without that space, the lipid hypothesis in its original form is logically disproven.
This is the only model that would explain how LDL-cholesterol particles might be able to “slip” — Subbotin uses the word “crawl” (11) — easily through a damaged endothelial lining to enter the acellular subendothelial space, where they are engulfed by invading macrophages, thus initiating the process of progressive atherosclerosis. But if this (hypothetical) acellular subendocardial space is not acellular but instead comprises layer upon layer of mature cells, how will the LDL-particles find their way between those cells? And how, among all those layers of cells, will the macrophages be able to locate the LDL invaders?
Subbotin cites the utterly fundamental and completely ignored work of Yutaka Nakashima et al. (12-14), which reveals two essential findings that destroy Keys’ lipid hypothesis. Interestingly, Nakashima and colleagues avoid any reference to the possibility that their work disproves Keys. Only Subbotin has had the courage to advance this heretical possibility.
The first finding is that histological examination of adult coronary arteries shows the tunica intima does not comprise a single layer of endothelial cells sitting atop an acellular empty space that exists, just waiting to be filled by LDL-cholesterol and engulfing macrophages. This is shown in Figure 3.
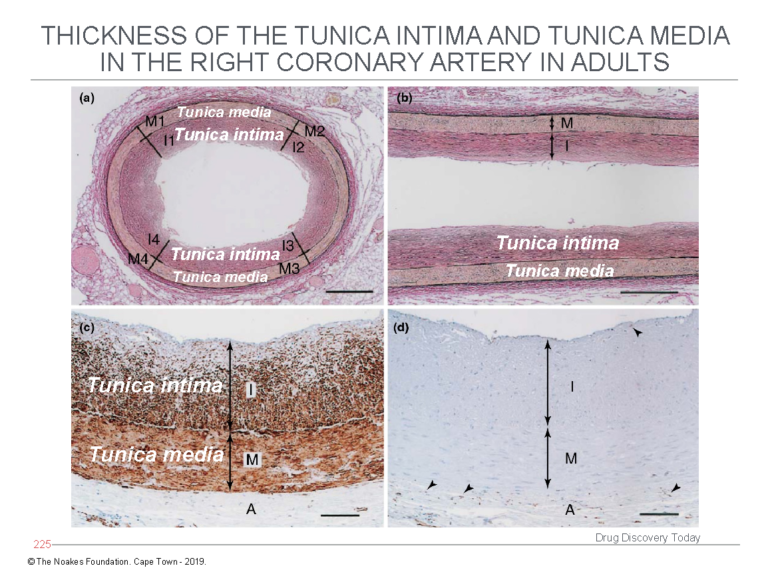
Figure 3: The study of Nakashima et al. (12) shows the tunica intima is not an empty acellular space as depicted in Figures 1 and 2. Instead, the tunica intima comprises multiple layers of cells (panels a and b) and is in fact thicker than the tunica media. Using a special stain to identify smooth muscle cells, panel c shows the cells in the tunica intima, below the endothelial layer, are indeed smooth muscle cells. Panel d stains for the presence of macrophages, which are identified with an arrow head. This evidence shows the diagrams depicted in Figures 1 and 2 are fundamentally incorrect in depicting the tunica intima as an acellular space potentially full of macrophages. Reproduced from Figure 3 in reference 10.
In contrast, the tunica intima comprises multiple layers of smooth muscle cells — up to 50 such layers — and without any macrophages.
The second critical finding reported by Nakashima and colleagues already in 2007 (13) and essentially forgotten until its rediscovery by Subbotin (10, 11) is the initial deposition of lipid material in the walls of arteries affected by atherosclerosis. This occurs in the deep layers of the tunica intima. These layers are separated from the endothelial cell layer by numerous layers of smooth muscle cells (Figure 4).
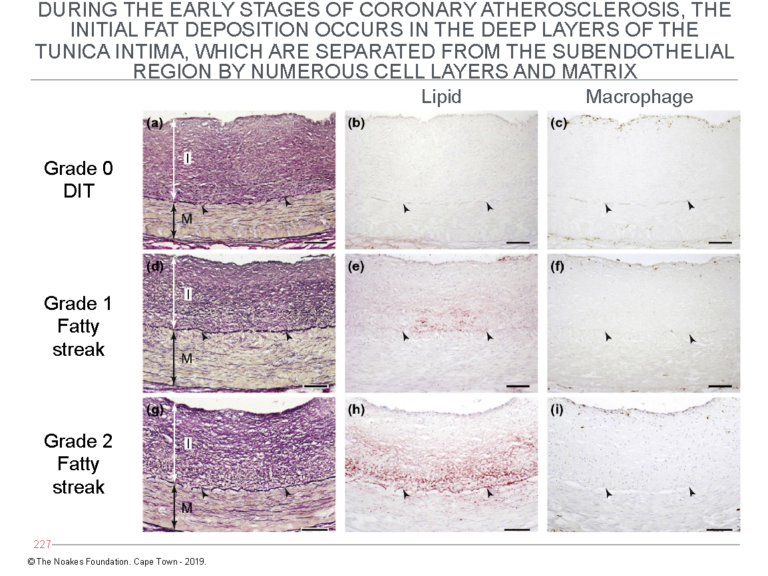
Figure 4: The panels on the left (panels a, d, and g) show the histological evolution of the fatty streaks in arteries of different subjects dying from different causes. The middle panels (b, e, and h) show the site at which lipid (staining red) begins to accumulate. Note especially in panel h that the main site of accumulation is in the deep layers of the tunica intima, close to the internal elastic lamina. The panels on the right (panels c, f, and i) are stained to detect the presence of macrophages. The panels show that despite some degree of lipid accumulation (panels e and h), there is no evidence for the invasion of macrophages. Figures 1 and 2 require that an invasion by macrophages into the (non-existent) subendothelial space is essential for the development of atherosclerosis. Reproduced from Figure 6 in reference 10.
The point of Figure 4 is to show that since the first evidence for lipid accumulation in diseased arteries occurs so deep in the tunica intima, it cannot have arisen from the LDL-cholesterol carried in the lumen of the arteries. There has to be another source for this lipid accumulation.
Figure 5 shows the progression of atherosclerosis with further lipid accumulation in the deep layers of the tunica intima. It also depicts the addition of macrophages in the latter stages — Grade 3 — of the progression of atherosclerosis.
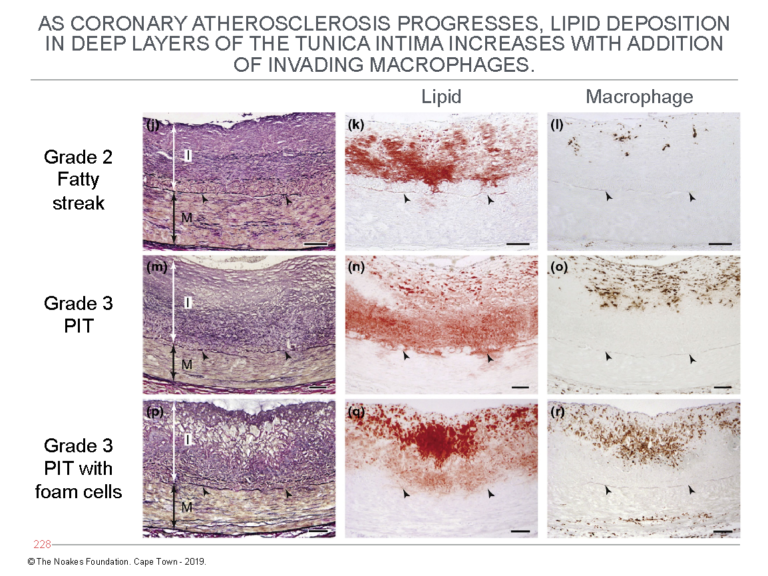
Figure 5: This figure shows the further progression of atherosclerosis from Grade 2 fatty streak to Grade 3 PIT with foam cells. It shows lipid accumulation increases in the deep layers of the tunica intima (central column of panels) with the addition of macrophages (column of panels on the right). The column of panels on the left shows the histological changes corresponding with this lipid accumulation in the deep layers of the tunica intima. Reproduced from Figure 6 in reference 10.
Figures 4 and 5 clearly establish that cholesterol circulating within arteries cannot explain why the atherosclerotic plaque begins to develop deep within the highly cellular tunica intima, far removed from where LDL-cholesterol is circulating in the bloodstream.
The sole conclusion must be that Keys’ lipid hypothesis cannot explain these findings. Hence, these findings disprove the essential foundations on which Keys’ theories are based.
Subbotin has proposed an alternate hypothesis (10, yet to be fully tested).
He proposes the fundamental event leading to the development of atherosclerosis is a triggering of proliferation (multiplication and growth) of the smooth muscle cells in the tunica intima. These are the cells in the arterial system that are known to replicate the most. Their replication can be initiated by any of a number of stimuli, including aging, transplantation, needle puncture, irradiation, hypertension, and some pharmaceutical drugs (10).
Subbotin postulates that following the triggering of their proliferation, perhaps by initiating stimuli yet to be fully understood, the mass of these cells increases. But a point will be reached at which this enlarged mass of cells can no longer remain viable without the addition of a dedicated blood supply (Figure 6).
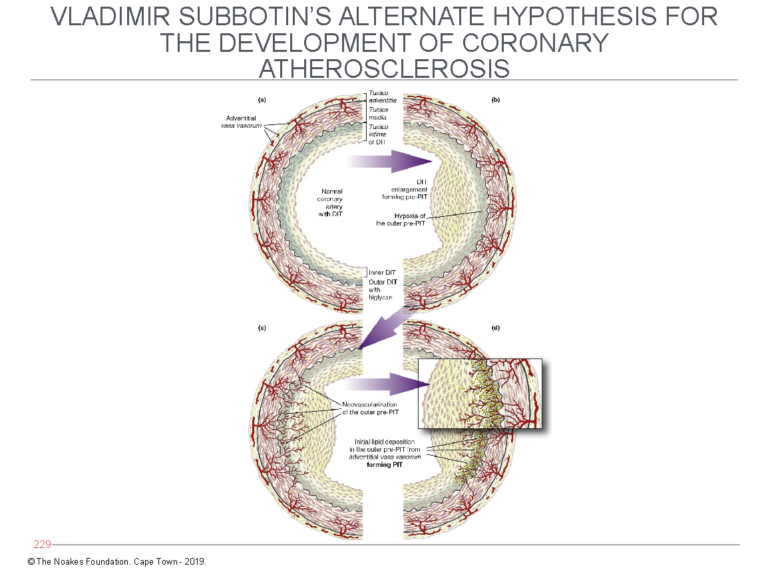
Figure 6: Subbotin has proposed that the normal coronary artery may develop diffuse (tunica) intimal hypertrophy (DIT) in response to currently unidentified stimuli. The result is that the cells in the outer layer of the tunica intima, furthest from the arterial lumen and their source of oxygen, become oxygen-deficient (hypoxic). The consequence is that new blood cells grow into the intima (left side of bottom panel) from the vasa vasorum. Blood entering the intima then deposits LDL-cholesterol, which explains how lipids enter the deep layers of the intima as depicted in Figures 4 and 5. Reproduced from Figure 7 in reference 10.
When that happens, the deepest layers of the intima recruit the development of new blood vessels (neovascularization). These blood vessels arise from the vasa vasorum, which exist in the tissue layer outside the tunica media and normally provide blood (and oxygen) to the muscle cells in the tunica media of muscular arteries.
Subbotin hypothesizes that once these new blood vessels enter the deepest layers of the tunica intima, they bring with them LDL-cholesterol, which is then deposited in that cell layer, producing the changes depicted in Figures 4 and 5.
Importantly, there is substantial evidence that the vasa vasorum are intimately involved in the development of atherosclerosis, and “present data indicate that vasa vasorum neovascularisation and atherosclerosis are seemingly inseparably linked” (15, p. 878).
In a presentation for CrossFit Health (11), Subbotin makes another extremely relevant point. He states that while lowering the blood cholesterol concentration with statin drugs may produce marginal health benefits in some, it does not cure the disease.
In fact, something like two-thirds of patients with coronary heart disease — the “forgotten majority” — will still suffer major coronary events despite using cholesterol-lowering drugs to normalize their blood cholesterol concentrations (16, 17). Although Harvard cardiology professor Peter Libby questions whether this is because treatment started too late, was of too short a duration, or was “too little,” Subbotin suggests that perhaps the hypothesis on which this treatment is based is simply false.
Subbotin quotes Claude Bernard: ‘‘Indeed, proof that a given condition always precedes or accompanies a phenomenon does not warrant concluding with certainty that a given condition is the immediate cause of that phenomenon. It must still be established that when this condition is removed, the phenomenon will no longer appear” (18).
In other words, because two phenomena are related in time (by association), this does not prove that one causes the other. For proof of causation, elimination of the supposedly causative factor must be followed by elimination of the condition being studied. Or, as William Stehbens has written, ‘‘Differentiating between cause and non-causative factors is essential. … Reduction in incidence rather than elimination of the disease precludes a causal relationship’’ (19).
In other words, unless removal of the supposedly causal agent — in this case, an elevated blood cholesterol concentration — produces a 100% cure in all persons, then the elevated blood cholesterol concentration cannot be the cause of the condition — in this case, coronary atherosclerosis and coronary heart disease.
That lowering the blood cholesterol concentration has not produced a 100% cure of coronary heart disease is clearly expressed in the frustration of Professor Ira Tabas in his 2016 Russell Memorial Lecture in Vascular Biology (20): “How can the overall lowering of plasma LDL over the past 3 decades, that is, after the introduction of statins, be reconciled with the fact that atherosclerotic vascular disease remains the leading cause of death (21)” (20, p. 187).
Unfortunately, he then exhibits the bias expressed by all who accept Keys’ lipid hypothesis without question: “Despite the enormous life-saving success of statins, issues related to … patient compliance, and patient and provider education have limited our ability to lower LDL to the types of level, and at an early enough age, that would be needed to remove atherosclerotic disease from the leading killer list” (20, p. 187).
In other words, “we” the cardiologists and researchers who accept the dogma of the lipid hypothesis, “know” that cholesterol-lowering is life-saving. But when we have evidence that it is not, the blame clearly lies with the patient and her health provider who simply did not lower her blood cholesterol concentration early enough and keep it low enough to produce the life-saving outcomes “we know” must naturally happen.
The possibility that the lipid hypothesis is false is never considered. That is simply an intellectual step too far.
But the true extent of the failure of the lipid hypothesis is summed up in the writing of Joseph Goldstein and Michael Brown, who, as I described earlier (22), were awarded the 1985 Nobel Prize in medicine or physiology for their discovery of the receptor mechanism by which cholesterol is transported into cells. Their discovery assisted in the launch of cholesterol-lowering drugs — statins — and the spread of hyperbolic claims that these drugs would eliminate coronary atherosclerosis and coronary heart disease.
Brown and Goldstein predicted in 1996 that “proof of the cholesterol hypothesis, discovery of effective drugs, and better definition of genetic susceptibility factors — may well end coronary disease as a major public health problem early in the next century” (23, p. 629).
That this has not happened (21) despite these hubristic predictions is perhaps the very best evidence that Keys’ lipid hypothesis is quite simply wrong.
What Else Is Not Consistent With This Traditional Explanation of How Atherosclerosis Develops?
Keys’ acolytes never acknowledge that primary outcome measures in epidemiological studies such as strokes and heart attacks, both fatal and non-fatal, are not an exact measure of the extent of coronary atherosclerosis in those populations (19). Rather, as E. B. Smith explains, they are a measure of acute thrombotic events that begin when the arterial plaque ruptures:
Thus the end-point of epidemiological studies and intervention trials is a thrombotic event, but most [researchers] have concentrated only on lipids and lipoproteins, and have made no measurement at all of blood homeostatic factors. However … several prospective studies of the relation between haemostatic function and myocardial infarction have been published, notably the Northwick Park (24) and Framingham (25) studies, and these have consistently shown that an increased level of fibrinogen is at least as significant a predictor as cholesterol. Furthermore the level of fibrinogen and activity of Factor VIII are increased in hyperlipidaemia (26, 27) so that there is a complex interaction between plasma lipids and thrombosis. (28, p. 235)
Smith concludes that fibrin deposition plays a major role in the development of atherosclerosis. But Keys’ hypotheses do not allow for this mechanism.
The practical point is that heart attacks and strokes can occur in persons with minimal atherosclerosis, whereas others with extensive disease may not ever develop an acute event. Even on an individual basis, it is not possible to predict which coronary artery plaques, big or small, might rupture. Indeed, small plaques are as likely to rupture as larger ones. This helps explain why the popular and highly lucrative cardiological practices of coronary bypass surgery (29) and coronary artery stenting do not produce beneficial outcomes in most conditions for which either is used (30-32). At least some of the benefit of stenting (and so perhaps also of coronary artery bypass grafting) is due to a placebo effect (33).
The reason is because arteries with minimal atherosclerosis are not considered candidates for the placement of a stent. However, the absence of severe atherosclerosis in a particular artery does not guarantee the complete absence of risk of plaque rupture in that artery, however minor the disease may be.
For similar reasons, coronary artery bypass grafting may be no more beneficial than standard medical care for the majority of patients with CHD (29).
So dominant has been the lipid hypothesis that there has been little interest in the role of different diets in promoting or limiting the probability of plaque rupture with resultant blood clotting (coronary thrombosis).
1910: William Procter and James Gamble Produce Novel Hydrogenated Seed Oil, Subsequently Called Crisco
William Procter came to Cincinnati, Ohio, in the 1830s after his candle-making business in England had been destroyed by fire. There he met candlemaker James Gamble, who had left Ireland during the Great Famine of the 1840s. When they married sisters, the brothers-in-law decided to combine their expertise and formed the company, Procter & Gamble (P&G).
At the time, Cincinnati was known as “Porkopolis,” as it was the center for the distribution of pork products down the Mississippi to New Orleans and the southern coastal United States. Pork fat was a key ingredient for P&G’s business; the pork provided the fat the company needed to produce soap and candles.
A recession in the 1870s forced the brothers-in-law to search for a competitive advantage. They decided to produce their soaps in individually wrapped bars, but for this they needed a cheaper source of fat. They settled on a mix of palm and coconut oils and created Ivory, the first soap that floated in water.
Their next challenge occurred in 1882 when Thomas Edison invented incandescent electric lighting (the electric light bulb) and formed the Edison Electric Illuminating Company of New York. Electric lighting spread quickly across the U.S., and Edison’s invention posed a very serious challenge to Procter & Gamble’s candle-making business. Their response would impact the health of future generations, right up to the present, in ways no one could then possibly have imagined.
To survive, P&G began to search for an alternative business. The brothers sought to turn their ability to produce soap and candles from rendered fats into some other product that would be widely used. They began by searching for a cheaper source of fat (oil) sourced from seeds, and they built a series of crushing mills to extract oil from cotton seeds.
American chemist David Wesson had by then perfected a method to bleach and deodorize cottonseed oil, making the extract clear, tasteless, and odorless (34). The oil was then sold as a liquid or, when mixed with animal fat, as a cheaper shortening that resembled lard. The composite fat became the chief competitor for lard used as shortening, especially in baking.
Soon the P&G chemists became interested “in the possibility of converting cottonseed oil into a solid form (to replace animal fats) for use in shortening … . They hoped this (novel compound) could compete in quality and price with lard, butter, and the many compounds already on the market” (35). But they needed a method to convert cottonseed oil into a more solid, less liquid form (like butter, lard, or tallow).
In 1907, German chemist E. C. Kayser contacted P&G (35). He had developed a process by which liquid cottonseed oil could be converted to a solid fat. The process he discovered is now known as hydrogenation. P&G invited Kayser to the U.S. Impressed by his story, the company purchased the rights to his process and hired him as a company consultant.
Three years later, on Nov. 10, 1910, P&G applied for a U.S. patent for a new “vegetable” (actually seed) shortening produced by Kayser’s hydrogenation process. The shortening consisted “of a vegetable (sic) oil preferably cottonseed oil, partially hydrogenized [hydrogenated], and hardened to a homogenous white or yellowish semi-solid closely resembling lard. The special object of the invention is to provide a new food product for a shortening in cooking” (35). The product was called Crisco. By 1912, it was being advertised as “an absolutely new product — a scientific discovery which will affect every kitchen in America” (35).
The immediate challenge facing P&G was that American housewives associated cotton seeds with clothing, not food. To circumvent this problem, the true source of the product was never disclosed (34). Instead P&G’s chief marketing points were that Crisco was cheaper than lard, and unlike lard, it was also tasteless. In addition, the more hydrogenated the “vegetable” oil, the less likely it was to melt on warmer summer days (in the days before the development of the refrigerator). This provided a particular advantage when used as shortening for baking in place of lard or butter, which melt at lower temperatures.
Even better, it was a novel product created by a novel chemical process (hydrogenation) by a company everyone could trust — all features that would appeal to the progressive American housewife of the 1920s. Using the new product would show how thoroughly modern she was, the company’s advertisers enthused. They added the unsubstantiated claim that this new product, created by modern technology, was also much healthier than the butter humans had been eating for tens of thousands of years. They carefully avoided the point that there was (and still is) no evidence that butter is anything other than extremely healthy for humans.
No one bothered to ask the obvious question: Why should a recently developed artificial product formed through a complex series of chemical reactions including bleaching, deodorizing, and hydrogenation be healthier for humans than foods like butter, lard, and tallow, which our ancestors evolved to eat over millennia?
Still today, no one asks that question. Unfortunately, 50% of the fat content of Crisco comprises trans-fatty acids, and it wasn’t until nearly a century after the invention of Crisco that the health dangers of trans-fatty acids became fully appreciated.
Skillful marketing ensured the product was an immediate success. Sales increased nearly 30-fold during the first four years of its production (36, p. 88), from 2.6 million pounds of Crisco in 1912 to 60 million pounds four years later.
The displacement of traditional sources of added dietary fat by these industrially produced, vegetable-based oils progressively increased (Figure 7), introducing perhaps the single largest dietary change in the U.S. (and elsewhere) since the beginning of the 20th century.
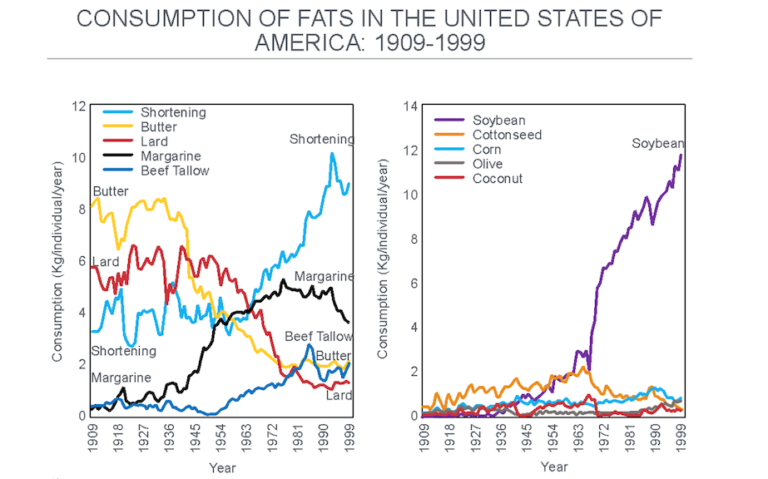
Figure 7: The change in the important sources of added dietary fat after 1909. Note the progressive reduction in the consumption of butter and lard as they were replaced with shortening and margarine, both produced by the hydrogenation of seed (“vegetable”) oils. The main seed oil now used in this process is soybean oil, which overtook cottonseed oil after about 1966 (right panel). Reproduced from reference 37.
Figure 8 shows what this change means in terms of the number of grams of the different added fats eaten daily by individuals in the U.S. The key difference is the replacement of saturated fat from butter and lard with shortening, margarine, and oils now derived from seed oils, which are rich in trans fats and omega-6 polyunsaturated fats (PUFAs).
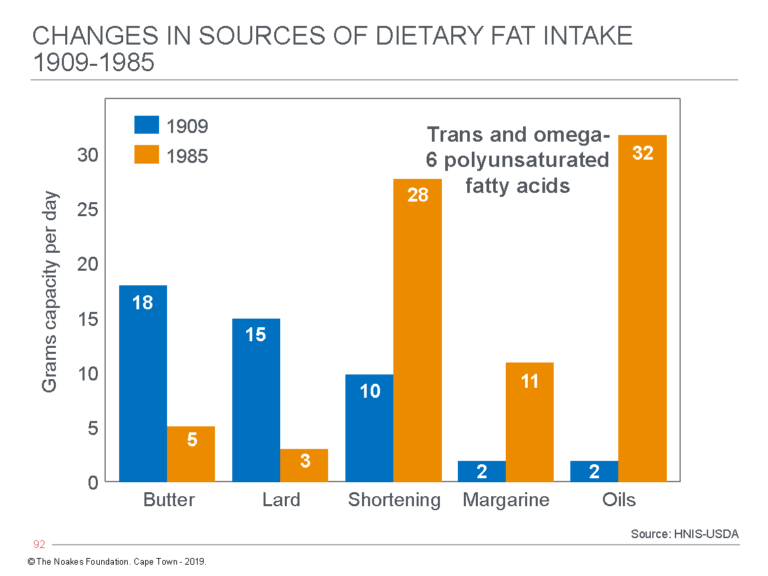
Figure 8: The change in average grams of fat consumed daily from butter, lard, shortening, margarine, and seed oils by individuals in the U.S. from 1909 to 1985. (Note that Crisco was marketed for the first time in 1910). In 1909, butter, lard, and shortening (then also derived from animal sources) provided 43 (91%) of the 47 grams of added fat eaten daily by U.S. citizens. Today the total intake from these sources is 79 grams, of which butter and lard now provide just 8 grams (11%), whereas processed seed oils in shortening, margarines, and oils provide 71 (89%) grams. Some argue this dietary change, rather than an overall increase in refined carbohydrate consumption, is the real cause of the rise in the modern diseases of “lifestyle” — actually diseases of nutrition. Redrawn from data from reference 38, p. 43.
While a great marketing ploy, P&G’s original 1910 claim that partially hydrogenated seed oils are healthier than animal fats was never based on any experimental evidence. With time, it simply became the accepted dogma according to the conclusion that any other fat, however produced, is healthier for humans than is saturated fat derived from animal products. To prove this claim, all the “healthier” fat had to do is lower the blood cholesterol concentration. Whatever other effects an industrially produced “vegetable” oil might occasion, good or bad, are utterly irrelevant according to this popular logic.
Interestingly, Keys clearly had some misgivings about the ill effects of hydrogenated fats, for already in 1956, he wrote:
A reasonable, practical conclusion from the present evidence might be to propose for American adults a sharp reduction in the total dietary fat from their current average intake in which fats account for some 40 per cent or more of the total dietary calories. In this dietary adjustment, emphasis might be placed on reducing the consumption of margarine, hydrogenated shortenings, butterfat, and meat fats. The great nutritional values of milk and meat would be maintained, and increased, by favouring skim milk, cottage cheese, and lean meat. (39, p. 376, my emphasis)
In 1961, he and his colleagues published a study showing hydrogenated “vegetable” oils containing a substantial amount of trans fats significantly increased blood cholesterol and phospholipid and triglyceride concentrations (40). Yet remarkably, Keys never again raised the alarm about the potential hazard of replacing dietary saturated fats with polyunsaturated hydrogenated fats containing substantial amounts of trans fats.
Understandably in 1910, the P&G chemists did not appreciate that the hydrogenation process produced this range of novel fats including trans fats. The extent to which trans fats are produced depends on the seeds being hydrogenated. With the processes used in the 1960s, 25% of the hydrogenated fats in corn oil were trans fats; in cottonseed it was 35%, in soy 40%, and in canola 50% (41).
1913: Animals Fed Cholesterol in High Doses Develop What Looks Like Atherosclerotic Arterial Disease
In 1913, the Russian scientist Nikolai N. Anichkov (spelled “Anitschkow” in the German literature) and his medical student S. Chalatow “purified cholesterol from egg yolks, dissolved it in sunflower oil, and fed it to normal rabbits” (5, 42). This caused the rabbits’ blood cholesterol concentrations to rise to the extraordinarily high value of 500-1,000 mg/dL (13-26 mmol/L), which is perhaps predictable since rabbits are herbivores that are not designed to eat animal products. Recall that only animal products contain cholesterol. When force-fed cholesterol, rabbits store the excess cholesterol in all their body tissues.
Anichkov and Chalatow were influenced by other research being undertaken by their colleagues at the St. Petersburg Imperial Military Medical Academy (43). Their studies found that rabbits raised on diets high in milk, eggs, and meat developed arterial lesions. Egg yolks, but not egg whites, caused the arterial lesions. Rabbits fed cholesterol extracted from egg yolks and dissolved in vegetable oil developed arterial lesions, superficially resembling human atherosclerosis. The authors concluded: “Since the same (arterial) changes can be observed by feeding pure cholesterol, there remains no doubt that it is precisely this substance that is laid down in the organism as liquid-crystal droplets and evokes extraordinarily damaging effects in various organs” (42, 43).
Anichkov’s findings were largely ignored when others failed to reproduce them in other laboratory animals, in particular in rats and dogs; the results were reproduced with only variable success in pigs and primates (38). The strongest criticism of his work remains that this form of experimental atherosclerosis produced by feeding animal products to herbivores is not the same as that found in humans (44). In addition, the potential role of oxidized cholesterol and diets deficient in essential fatty acids in causing arterial damage in some of these studies was not appreciated (38). Despite these failings, Anichkov’s studies were resurrected in the 1980s by those wishing to find a more solid foundation for the increasingly shaky lipid hypothesis.
Daniel Steinberg, MD, who built his medical legacy on an unflinching support for Ancel Keys (45, 46), later wrote:
Anitschkow’s body of work showed clearly and convincingly that hypercholesterolemia in rabbits was a sufficient cause of atherosclerosis. Of course it did not necessarily follow that cholesterol — either in the diet or in the blood — was also an important factor in human atherosclerosis. That conclusion would have to await studies showing that hypercholesterolemia in humans was indeed associated with atherosclerosis and, ultimately, clinical trials to establish that relationship as a causal one. (5, p. 2948, my emphasis)
This, it should be emphasized, is proof by circular argument. If Anichkov’s work had “clearly and convincingly” established that high blood cholesterol concentrations are the direct cause of human atherosclerosis, then there would be no need for any more studies, including clinical trials in humans to “establish that relationship.” In science, the truth stands by itself. A theory does not become the truth when bolstered by yet another still unproven theory. This is the lesson Keys and his acolytes failed to learn.
The clinical trials to which Steinberg refers failed spectacularly in establishing that an elevated blood cholesterol concentration is the key or even a necessary factor in the development of atherosclerosis.
Following the logic of Steinberg’s circular argument, if those clinical trials are inconclusive, then Anichkov’s finding is equally meaningless — at least as it relates to human atherosclerosis. But over the years, the most vociferous advocates of that unproven relationship have, like Steinberg, been those whose careers have been the most dependent on the relationship being true.
The campaign to make cholesterol the principal villain in causing atherosclerosis began in earnest in the U.S. in the mid-1980s. It started with the 1984 NIH Consensus Development Conference (NIHCDC), chaired by none other than Steinberg himself. The principal outcome of the NIHCDC was the National Cholesterol Education Program (NCEP), the main focus of which was to convince the majority of U.S. physicians who at the time were skeptical that cholesterol causes heart disease — so-called “cholesterol skeptics” — that their skepticism was misplaced.
Central to all of this was the activism of Steinberg himself. Clearly, the man driving the Consensus Development Conference had first to be convinced of the correctness of his theory, but that was not difficult as Steinberg, like Keys, was never in any doubt:
Today, we know that we are winning the war against coronary artery disease. In fact, clinical trials with the statins have shown remarkable decreases in both coronary heart disease mortality and also total mortality. Decreasing LDL by ~25% is enough to lower coronary heart disease mortality by 30-40%, and that is the result of only 5 or 6 years of intervention. It seems reasonable to extrapolate and expect even greater reductions if treatment is started earlier in life and continued not for just 5 years but for decades. (46, p. 1585)
Later he would add: “Yet the validity of the lipid hypothesis did not become generally accepted until 1984. (Believe it or not, there are still a few pockets of stout resistance!)” (5, p. 2949).
Steinberg’s militaristic language (recall his book was also titled The Cholesterol Wars) shows he believed the crusade to market cholesterol as the demon causing heart disease was never simply a scientific process. It had become a war, rife with confirmation bias. Personal egos, funding opportunities, and academic reputations had to be protected at all costs.
In the end, those costs would be borne by the citizens of the world as they suffered through the global obesity/diabetes epidemic this “war” initiated. It is as British Prime Minister Neville Chamberlain said: “In war, whichever side may call itself the victor, there are no winners, but all are losers.” The cholesterol wars are no exception.
Those who disagreed with Steinberg’s fake consensus quite naturally became the hated enemy who deserved to be exterminated. Two were quickly dispatched: John Yudkin, MD, as I described previously (48), and George Mann (49), the co-founder of Framingham Study who later described Keys’ diet-heart hypothesis as the greatest health scam of the (20th) century:
The hypothesis, proposed in the late 1950s, that atherosclerosis is caused by the saturated fatty acids and cholesterol in our diet was based upon scanty, imperfect and highly selected information. That hypothesis has been repeatedly tested over the last 40 years in dozens of extraordinarily expensive and complicated trials. In every instance when the outcomes are honestly interpreted, the results have been the same. No dietary modification and no drug treatment has been shown to correct the problem of CHD. The diet-heart hypothesis has been repeatedly shown to be wrong, and yet, for complicated reasons of pride, profit and prejudice, the hypothesis continues to be exploited by scientists, fund-raising enterprises, food companies and even government agencies. The public is being deceived by the greatest health scam of the century. (50, p. 1)
As a reward for his opposition, Mann’s career was essentially terminated. As he told Nina Teicholz: “‘It was pretty devastating to my career … . One day,’ recalls Mann, ‘the woman who was the study section secretary asked me to step out in the hall. ‘Your opposition to Keys is going to cost you your grant,’ she said. And she was right’” (36, pp. 67-68).
1945: CHD Mortality Falls in Some European Countries During World War II But Not U.S.
A series of articles from European authors (51-57) reported that deaths from CHD fell quite sharply during the latter years of World War II. This was typically associated with a reduced consumption of animal products “rich in cholesterol” — particularly meat, whole milk, cream, cheese, and eggs — alongside increased consumption of carbohydrate-rich foods like cereals, potatoes, and fruits. Although sugar consumption also fell sharply, it was the reduction in the consumption of animal produce and cholesterol that caught the attention of these authors.
As a result, Haqvin Malmros (51) produced the first iteration of what would become known as Keys’ diet-heart and lipid hypotheses: “We ought to avoid all luxury consumption of high-cholesterol foodstuffs. Especially the combination of eggs and cream, butter or other fats would seem to be risky … . There is much which goes to show that in Denmark, Sweden and the U.S.A. the consumption of eggs, butter, milk and other animalic fats is at present too high and may involve serious risk for public health” (51, pp. 152-153).
The risk of drawing this conclusion was that many factors in the lives of these civilians changed as a result of the war. Such a conclusion about nutrition requires an a priori assumption that nutrition was indeed the factor that produced the observed health changes. There is a saying in science that what you believe determines what you believe. The scientific term for this is confirmation bias.
After the war, evidence became available in Norway that CHD rates were again on the rise and were associated with an increased intake of dietary fats (58). Interestingly, intake of dietary trans-fatty acids increased dramatically as the Norwegian government subsidized margarine in the post-war years, and this margarine contained trans fats in high concentrations.
April 1945: President Roosevelt Dies From Fatal Stroke
The 31st President of the U.S., Franklin Delano Roosevelt, was known to suffer from elevated blood pressure, rising from a value of 136/78 mmHg in 1935 to 188/105 mmHg in 1941 (90, p. 2). At the time, the president’s personal physician reported his blood pressure was “no more than normal for a man of his age” (90, p. 2), but others were less certain. British Prime Minister Winston Churchill asked his personal physician, Lord Charles Moran, whether he too had “noticed that the president is a very tired man” (59, p. 2).
In March 1944, Roosevelt was admitted to the Bethesda Naval Hospital for evaluation due to abdominal distension and increasing shortness of breath upon exertion. There, cardiologist Howard Bruenn diagnosed hypertension, hypertensive heart disease, and cardiac failure. The treatment then available for the president’s condition was primitive and largely ineffective. A month later, Roosevelt’s blood pressure had risen to 240/130 mmHg. Today, that would be diagnosed as malignant hypertension requiring the patient to be hospitalized without delay.
At the Yalta Conference in February 1945, it was clear to all that the president was gravely ill. Lord Moran wrote in his diary, “The President appears a very sick man. He has all the symptoms of hardening of the arteries … . I give him only a few months to live” (59, p. 3).
In April 1945, as the Allied armies fought their way into Germany, Roosevelt’s health had deteriorated noticeably. He was weak with an ashen-gray complexion, features in keeping with advanced heart failure. To recuperate and regain his strength, he traveled to the “Little White House” in Warm Spring, Georgia, but on April 12, 1945, at 1:15 p.m., the president, then 63, suffered a massive cerebral hemorrhage (stroke), dying two hours later. His blood pressure was 300/190 mmHg (60).
This anecdote makes it clear that in 1945, very little was known about the effective management of hypertension, hypertensive heart disease, and cardiac failure. Nor was the role of hypertension as a “risk factor” for CHD and hypertensive heart disease widely known.
This level of ignorance, exposed by the president’s death, became perhaps the single greatest driver of medical research in modern times. The goal was simple: Understand what causes coronary heart disease so the disease can be prevented in the future.
1948: President Harry Truman Signs the National Heart Act Into Law
The death of a sitting U.S. president from a cerebral stroke captured the attention of Congress, which soon agreed to support research that would improve the understanding, treatment, and prevention of heart disease.
The result was that on June 16, 1948, President Harry Truman signed into law the National Heart Act. The act stated: “Congress hereby finds and declares that the Nation’s health is seriously threatened by diseases of the heart and circulation, including high blood pressure … . These diseases are the main cause of death in the United States and more than one in every three of our people die from them.”
The new law established the National Heart Institute (NHI), today known as the National Heart, Lung, and Blood Institute (NHLBI), and granted $500,000 in seed money for a 20-year epidemiological heart study. That study became the Framingham Heart Study.
1948: Procter & Gamble Donates $1.74 Million to the American Heart Association (AHA)
The American Heart Association (AHA) began as a non-government organization, underfunded and with “virtually no income” (23, p. 47), and so it might have remained if it weren’t for the fact that in 1948, as Teicholz describes, “It got lucky: Procter and Gamble (P&G) designated the group to receive all the funds from its ‘Truth or Consequences’ contest on the radio, raising $1,740,000, or $17 million in today’s dollars” (36, pp. 47-48).
The result was that the AHA was able to hire its first professional director, “a former fund-raiser for the American Bible Society” (36, p. 48).
By 1960, the AHA’s fundraising efforts were so successful that it was able to invest hundreds of millions of dollars into heart disease research.
Thanks to P&G’s contribution, the AHA had become “the authoritative source of information about heart disease for the public, government agencies, and professionals alike, including the media” (36, p. 48). Indeed, the organization would grow to become one of the most trusted NGOs in the world.
And it never strayed far from the dietary recommendation to replace saturated fat with “vegetable” oils, some of which are conveniently manufactured by the foundation sponsor that made all this possible: P&G.