
The previous column finished with the story about how Ancel Keys and his disciples buried the data from the Minnesota Coronary Experiment (MCE). If the true findings of that study (2) had been released during his lifetime, Keys’ diet-heart and lipid hypotheses would have been dead and buried, and the world would have been spared the disastrous epidemics of obesity and Type 2 diabetes mellitus (T2DM) that resulted when Keys’ unproven hypotheses were accepted and marketed as facts.
But this did not happen, because the forces that Keys’ scientific activism had unleashed were simply too large and too powerful to be contained.
Instead, Keys’ hypotheses, disproven in 1975 by the results of the MCE, continue to drive the global nutrition agenda, also determining what medical and dietetics students are taught in their training.
The direct consequences are the ill health we see all around us.
In the subsequent column on Keys and his influence, I sketch the case of scientific misconduct of which I believe he is guilty (3).
But first in this column, I consider the implications of the results of two other outrageously expensive epidemiological studies that were influenced by his work.
The Framingham Heart Study inconveniently fails to support Keys’ hypotheses
A fact that I uncovered only by reading Keys’ personal memories of the Seven Countries Study (SCS) (4) was that when he applied for funding for what would become his Minnesota Business and Professional Men Study (5), his grant was rather too rapidly approved. The reason, at least according to the 90-year-old Keys (4, p. 16), was that the funding body at that time, the U.S. Public Health Service, liked his proposal so much that it decided to steal his idea and fund a more extensive study of its own. That study, located in the small town of Framingham, Massachusetts, just outside Boston, would become iconic. It was called the Framingham Heart Study (FHS) (6). The study officially began when it admitted its first subject on Oct. 11, 1948.
The goal of the study was to determine whether it is possible to identify risk factors that in any population will identify those who will develop one or another chronic disease, coronary heart disease (CHD) in particular. It’s important to understand that before the FHS, what would become known as coronary risk factors did not exist. Such risk factors owe their beginnings to the FHS. But did the FHS actually identify CHD risk factors that are of real practical value to the individual or the practice of clinical medicine? For the past 50 years, the medical profession has been directed to accept these risk factors as an article of faith. But the truth is rather more nuanced.
The results from the FHS began to be published concurrently with those from the SCS and MCE. The findings of all three studies were then marketed as definitive proof of Keys’ twin hypotheses (the diet-heart and lipid hypotheses). One result was the global drive to replace saturated fat in the diet with polyunsaturated fatty acids (PUFAs), specifically to lower blood cholesterol concentrations in order to prevent CHD.
As described in riveting detail by Dr. Michael Eades, MD, from the outset, the FHS was bedeviled by a legion of methodological and other problems — perhaps predictably because it was the first such study of its kind (7-9). In the end, these problems reduced the scientific value of the study.
The first problem was that it was just another observational, associational, non-randomized study (7). And as I detailed in the previous column (10), observational studies cannot prove causation, except in a few very exceptional circumstances (Figure 1, reference 10).
The second problem was the presence of participants who were judged to be “unhealthy” when they joined the study because they had elevated blood pressures or high blood cholesterol concentrations. How were they to be managed? What if they received treatment that would influence the study’s findings? Would those treatments be recorded and analyzed appropriately?
The reality is that prospective observational studies are only valid if they begin with a population that is sufficiently healthy to not require medical interventions from the start.
The third problem was to define what the original (baseline) blood cholesterol concentration was, because it seemed to vary rather more than anyone expected. Their experimental model (in which blood cholesterol concentrations are measured just once in the subject’s life) did not consider that perhaps blood cholesterol concentrations vary over time. Rather, real-life testing of Keys’ lipid hypothesis has always presumed blood cholesterol concentrations remain static without intervention. But persons originally found to have high blood cholesterol concentrations might, a few years later, have low concentrations without any discernible changes in their diets or lifestyles. Of course, the reverse might also apply.
This would mean the baseline blood cholesterol concentration might be of little value in predicting each individual’s blood cholesterol concentration over the duration of the trial.
Ironically, a study by Keys and his colleagues established substantial variability in individual responses to dietary changes designed to lower blood cholesterol concentrations so that “assessment of the real effectiveness of a dietary regimen in an individual is best based on observed dietary changes. Total cholesterol changes among individuals under treatment should be based on multiple determinations and interpreted with caution” (11, p. 349).
In trying to make sense of this circuitous logic, this means, I suspect, that a low-fat, low-cholesterol diet that is expected to prevent CHD by lowering the blood cholesterol may, in real life, still prevent CHD even if it does not lower (and might even raise) the blood cholesterol concentration — a conclusion that neatly contradicts the lipid hypothesis. But the authors, in their circumlocution, perhaps hoped the general reader would not have either the intellect, persistence, or desire to detect this.
Despite these massive, perhaps insurmountable problems, for the past 70 years, medical doctors and medical students across the globe have been taught that the FHS provided the key evidence supporting both the diet-heart and lipid hypotheses, as well as the use of coronary risk factors to identify those at future risk for developing CHD.
The reality is that most of what the study actually found has either been willfully misrepresented or suppressed.
In the FHS, dietary fat intake did not determine blood cholesterol concentration
An important goal of the FHS was to “prove” dietary fat intake determines blood cholesterol concentration: the very relationship Keys believed he had proven in his associational population studies (Figure 10 in reference 12). To study this, “Detailed medical histories were recorded on a sub-sample of about 1000 subjects by a carefully tested procedure. Ms. Georgeana Pearson spent about 3 years interviewing those people. It was a meticulous enterprise,” George Mann, MD, explained. Mann concluded, “The data clearly showed no relationship between dietary intakes of either fat or cholesterol and the subjects’ level of cholesterolemia or their experience of CHD” (13, p. 9, my emphasis).
Mann was initially one of the scientific leaders of the FHS but later resigned to express his displeasure at the fake science he detected. He noted: “These (dietary) data were never properly published because the findings were contrary to the position held by the National Heart, Lung, and Blood Institute (NHLBI). That organization runs the Framingham study. To withhold that information is a form of cheating. Nevertheless, the Framingham study, now a 40-year longitudinal study of risk factors for CHD, has failed to show that any measured dietary behaviour is a risk factor for CHD” (13, p. 9). As Mann told Nina Teicholz many years later: “That went over like a wet blanket with my superiors at NIH because it was so contrary to what they wanted to find … they wouldn’t allow us to publish that data” (14, p. 66-67, my emphasis).
In 1979, Keys’ co-workers, including Henry Blackburn, MD, admitted that in the Framingham study, “zero or near zero correlations were found between the various components of diet and serum cholesterol levels” (15, p. 77). They added: “Nichols et al. (16), in reporting these results from Tecumseh, state that the zero correlations ‘provide evidence that other factors besides fat intake are determinants of cholesterol levels among the general public’ and further that ‘from the findings in this study one may infer that weight reduction should be the initial intervention for control of hyperlipidemia in the general population” (15, p. 77). Furthermore, they concluded that epidemiological studies are not suitable for studying the relationship between dietary fat intake and blood cholesterol concentrations, another post-hoc explanation for an inconvenient research finding (Presumably, epidemiological studies are just fine if they provide the associational relationships that support the authors’ biases).
True scientists, whose goal is a search for the truth, do not invent such post-hoc explanations when the data they collect disproves their original hypothesis. They simply reject that hypothesis and begin again (17), formulating and testing a new hypothesis informed by the hypothesis that their carefully conducted experiment has just disproven.
Eades (9) confirms he has a personal copy of the FHS document that proves the FHS failed to show any relationship between dietary fat intake and blood cholesterol concentrations in the Framingham population. It was written by Tavia Gordon and was the 24th in the 28-volume official FHS report.
But the data were simply “lost,” buried deep within the vaults of the NIH archives in Washington (14, p. 67) until they too were inconveniently “recovered.”
This occurrence had much in common with the true data for the Sydney Diet Heart Study (18) and the MCE (2), which also mysteriously and conveniently “disappeared” only to be “recovered” many decades later.
Gordon discovered that when the diet records of persons with high blood cholesterol concentrations were compared with those who had very low concentrations, “they differed not at all in the amount or type of fat consumed” (19, p. 27). His report concluded: “There is a considerable range of serum cholesterol levels within the Framingham Study Group. Something explains this individual variation, but it is not diet (as measured here)” (19, p. 27).
Many years later, the director of the FHS wrote the following in an article published in a relatively obscure medical journal: “For example, in Framingham, Mass., the more saturated fat one ate, the more cholesterol one ate, the more calories one ate, the lower the person’s serum cholesterol” (20). Subsequently, he wrote, “Epidemiologic data on diet is a risky analysis because it seems that people who eat the most cholesterol and the most fat have the lowest blood cholesterol levels. People who eat the most calories seem to weigh less than those who eat the least. This is just the opposite of what one might expect. (21, p. 58)
But once more, the researchers, by this time too conflicted to reveal this truth, did all they could to ensure their disproved hypothesis survived fully intact.
Keys himself had already shown as early as 1950 (22), and subsequently confirmed in 1956 (23), that dietary cholesterol intake was unrelated to blood cholesterol concentration: “It is concluded that in adult men the serum cholesterol level is essentially independent of the cholesterol intake over the whole range of natural human diets. It is probable that infants, children and women are similar,” he wrote (23, p. 54).
And since animal products are the sole source of dietary cholesterol, this would tend to suggest that animal products cannot be the cause of atherosclerosis, at least according to Keys’ original hypothesis that it is dietary cholesterol that raises the blood cholesterol concentration, thereby causing CHD. This was a theory he would subsequently abandon in favor of the saturated-animal-fat diet-heart hypothesis.
Confirming this change, in 1991 Keys remarked, “Dietary cholesterol has an important effect on the cholesterol level in the blood of chickens and rabbits, but many controlled experiments have shown that dietary cholesterol has a limited effect in humans” (24, p. 584).
In 1987, Keys had also said, “I’ve come to think that cholesterol is not as important as we used to think it was. Let’s reduce cholesterol by reasonable means, but let’s not get too excited about it” (19, p. 79; 25).
The FHS also proved blood cholesterol concentration has no value in predicting risk of future CHD
The ability of elevated blood cholesterol concentrations to predict future risk of heart attack appeared very promising in the first six years of the FHS. As W. B. Kannel et al. explained, “Cholesterol levels tended to be higher in those who subsequently developed CHD in the six years of observation than in the population at risk. This elevation was most marked for men in the youngest age group and diminished with age” (26, p. 39, my emphasis). Analysis by group, they wrote, “reveals a gradient of risk of developing CHD with increasing levels of serum cholesterol, such that those with serum cholesterols over 244 mg per 100ml had more than three times the risk of CHD as do those with cholesterol levels less than 210 mg per 100ml” (p. 39).
As would become standard practice in this field, nowhere did the authors explain exactly what “more than three times the risk of CHD” actually means to the individual at risk. The claim that an elevated blood cholesterol concentration increased risk so profoundly allowed them also to justify this assertion: “That blood cholesterol is somehow intimately related to coronary atherosclerosis is no longer subject to reasonable doubt” (9, p. 7; 14, p. 65). By hyping the risk, the authors’ goal was to drive out doubt; to entrap the world into accepting their unproven hypotheses as facts.
In fact, the data showed the overall risk of developing CHD in the studied group was 3.6% for the six years of the study, or 0.6% per annum for each individual. Thus, a three-fold increase in risk meant that 1.8 of 100 men in the high-risk cholesterol group would develop CHD each year compared to only 0.6 in the group with the lowest blood cholesterol concentrations.
This suggests a blood cholesterol concentration elevated above 244 mg/100 mL (6.3 mmol/L) is not a particularly powerful predictor of who will develop a heart attack over the next 12 months. Annually, 98% of the men considered to be at the highest risk because they began that year with an “extremely high” blood cholesterol concentration would still be alive 12 months later. Clearly, something other than just the blood cholesterol concentration must also be involved.
In contrast, another early report from Thomas Dawber et al. (27) initially focused specifically on 898 men between 45 and 62 years old at the original examination; 52 of these men subsequently developed CHD in the first four years of the study. The authors found there was “no apparent gradient [for increasing CHD risk] below a [blood cholesterol concentration] of 260 mg/dL (6.7 mmol/L),” which was “well above the mean value of 225.5 mg/dL (5.8 mmol/L) for the entire group of Framingham men in this age range” (27, p. 1775).
This means for the vast majority of persons in the FHS whose blood cholesterol concentrations were below 260 mg/dL, all their low(-ish) blood cholesterol concentrations could predict was that they were not at any increased risk for CHD. This is rather different than the message coming from other original FHS reports (26, 28-30).
Compare it to the 3- to 3.6-fold increase in risk among women with impaired glucose tolerance in this same population (28, 29, 31). Thus, Kannel wrote, “a combination of obesity, low HDL cholesterol, and impaired glucose tolerance predisposes especially” (28, p.419). Was Kannel perhaps hinting that insulin resistance might be a better predictor of future CHD risk? (Recall that the factors he mentions are some of the key markers of insulin resistance.)
Interestingly, Kannel also reported: “Diabetic men — both those who did and did not develop atherosclerotic cardiovascular disease — had lower total cholesterol than non-diabetic subjects” (29, p. 1101). And: “Diabetic men … were more obese, had lower cholesterol, and smoked less than their non-diabetic counterparts” (31, p. 10). He also concluded: “The CHD risk in diabetic individuals cannot be explained entirely in terms of the usual CHD risk factors” (29, p. 1104); “A number of pathogenic possibilities deserve greater attention, including a possible contribution of hyperinsulinemia and possible adverse effects of hypoglycemic agents” (p. 1105); and, “The atherosclerotic process is complex, involving more than blood lipids” (p. 1105).
Yet he still signaled his rigid devotion to the omertà of the Keysians: “Atherosclerosis is a multifactorial process, with blood lipids intimately and fundamentally involved in its evolution” (p. 1105).
As the study progressed, the accuracy of an elevated blood cholesterol concentration as a predictor of future CHD risk became, at best, even more tenuous (1). Worse, half of all participants who experienced heart attacks had blood cholesterol concentrations below the normal level of 220 mg/dL (14, p. 65).
Although there was some relationship between elevated blood cholesterol concentrations and risk of CHD before age 50, after that age the relationship became inverse. K. M. Anderson et al. explained, “After age 50 years there is no increased overall mortality with either high or low serum cholesterol levels. There is a direct association between falling cholesterol levels over the first 14 years [of the FHS] and mortality over the following 18 years [so that for every 1 mg/dL drop in blood cholesterol levels, overall mortality increases by 11% and CVD death rate by 14%]” (1, p. 2176, my additions).
It is naturally impossible, according to Keys’ twin hypotheses, for blood cholesterol concentration to be both a risk factor for and a protective factor against CHD mortality depending simply on the subject’s age.
Keys’ model predicts high blood cholesterol concentrations cause a progressive worsening of atherosclerosis with increasing age. His model cannot possibly predict that the same elevated blood cholesterol concentrations that cause coronary atherosclerosis up to the age of 49 years and 364 days suddenly begin to remove all that established atherosclerosis the very next day. If the biology makes the finding look ludicrous (as this example clearly does), then the finding is an artifact of something else. It should demand that we begin the search for a different explanation. Unfortunately, because the diet-heart and lipid hypotheses are so entrenched, this has simply not been allowed to happen.
Visualizing The Framingham Data
In 1991, R. L. Smith and E. R. Pinckney (32) published their analysis of the Framingham data (28) provided by Kannel (Figure 1).
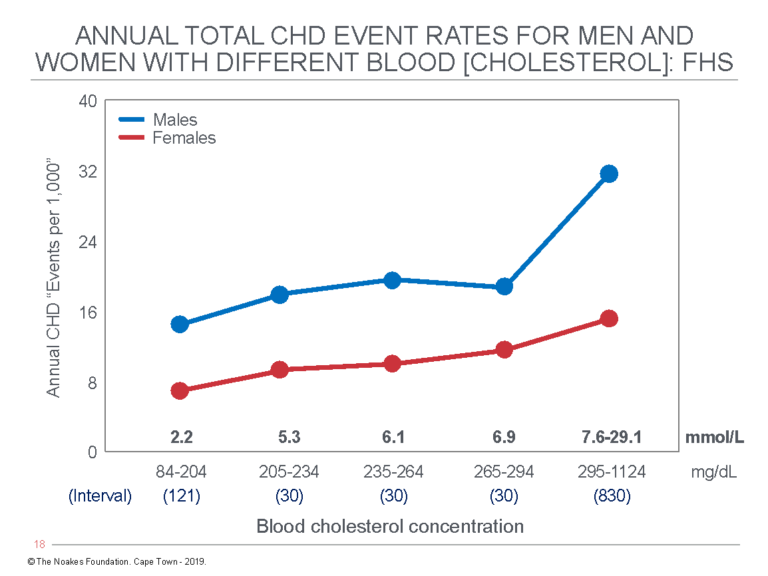
Figure 1: This figure shows the combined annual fatal and non-fatal CHD events for different blood cholesterol concentrations in men and women for the Framingham cohort (28). An important point is that the five blood cholesterol intervals in the original publication are unequal; the fifth interval includes blood cholesterol concentrations differing by 830 mg/dL (22.0 mmol/L) and comprises about 5% of the studied population. The apparently large increase in CHD events in men at blood cholesterol concentrations above 7.6 mmol/L is an artifact of this unequal division of blood cholesterol intervals above 6.9 mmol/L. Reproduced from reference 32, p. 38.
The key point in Figure 1 is that annual CHD event rates increase almost imperceptibly with increasing blood cholesterol concentrations in the range found in most humans (2.2 – 7.5 mmol/L). Thus, the annual CHD event rate predicted for persons whose blood cholesterol concentrations increase from 84 mg/dL (2.2 mmol/L) to 294 mg/dL (7.6 mmol/L) rises from 15 to 18 per 1,000 individuals, or from 1.5 to 1.8%. A 350% increase in the blood cholesterol concentration, then, increases annual CHD event rates in this population by 0.3%. Kannel considered this to indicate that “the serum total cholesterol is a powerful risk factor for CHD in both sexes” (28, p. 414, my emphasis).
Given the evidence he had at the time, Kannel can only have drawn that conclusion because of his blind devotion to Keys’ twin hypotheses, which is understandable since Kannel’s major life work was the FHS. And one driving goal of the FHS had become the support of Kannel’s buddy, Ancel Keys.
The Framingham data also confirmed blood cholesterol concentration could not predict the risk of sudden death. So in the end, Dawber had to concede: “The lack of association between serum cholesterol level and the incidence of sudden death suggests that factors other than the atherosclerotic process may be of major importance in this manifestation of coronary artery disease” (19, p. 62).
A number of other clinicians, some involved in the FHS, ultimately expressed their disappointment at this inability of blood cholesterol concentration to predict CHD risk:
- Kannel, one of the first directors of the FHS, wrote: “Diagnosis of overt heart disease on the basis of lipid (cholesterol) levels alone is not feasible” (33, p. 892), and, “At the levels of serum cholesterol noted in this population, however, cholesterol could not be demonstrated to be a necessary or sufficient cause of coronary heart disease. The disease must be looked upon as resulting from the interplay of multiple inter-related factors” (p. 896).
- FHS Director Dr. William Castelli wrote: “Obviously, the total cholesterol value cannot accurately predict which patients have a lipid problem when the cholesterol levels are between 200 and 250 mg/dL or even between 150 and 250 mg/dL. It is in these situations that the high-density lipoprotein cholesterol level becomes an important factor” (34, pp. 27-28).
- Castelli also wrote: “Few realize that half of the patients in whom coronary heart disease will eventually develop have cholesterol values below 250 mg/dL (6.5 mmol/L)” (34, p. 23). As Smith and Pinckney have explained, “Since the average cholesterol level among adult Americans is about 220 mg/dL, his statement means that heart attacks occur almost equally across all cholesterol levels” (32, p. 44).
- And Castelli again: “Those individuals who had total cholesterol (TC) levels of 150-300 mg/dL (3.9-7.8 mmol/L) fell into the overlapping areas … demonstrating that 90% of the TC levels measured were useless (by themselves) for predicting risk of CHD in a general population. Indeed, twice as many individuals who had a lifetime TC level of less than 200 mg/dL (5.2 mmol/L) had CHD compared with those who had a TC level greater than 300 mg/dL (7.8 mmol/L).” (35, p. S3). At this point, Castelli argued the additional measurement of “the three most easily assessed lipoproteins … HDL, LDL and VLDL” should also be measured to improve the risk prediction of the TC measurement. This was because he was beginning to understand that high HDL-cholesterol levels are associated with “a low risk of CHD” (35, p. S3). Thus, a series of graphs published in this paper showed an HDL-cholesterol concentration of >60 mg/dL eliminated any increased risk for CHD of a high TC, even >260 mg/dL (6.7 mmol/L). He also recognized plasma triglyceride concentrations “are an independent risk factor for CHD, even after making allowances for the HDL cholesterol concentrations” (p. S5). The next generation of FHS researchers would finally appreciate that insulin resistance can explain all these findings.
- Dr. Scott Grundy wrote: “The total number of coronary deaths is greater in the segment of the population with cholesterol levels below 250 mg/dL (6.47 mmol/L)” (36, p. 2855). Grundy also predicted: “A reduction in cholesterol level of 50 mg/dL (1.29 mmol/L) should cut risk of CHD in half” (p. 2855). Since cholesterol-lowering drugs reduce blood cholesterol concentrations substantially more, according to this prediction, heart disease rates should have been halved when statin drugs were introduced. This has not happened, so the basis for Grundy’s prediction is wildly inaccurate.
- Dr. George Livshits et al. (37) wrote: “The implications of our findings are that the evaluation of coronary risk should always include measurement of HDL-cholesterol percentage, especially in subjects with ‘normal’ (5.18 mmol/L) or only mildly elevated total cholesterol (<5.83 mmol/L), and in those with a total cholesterol of 5.83 to 6.47 mmol/L. In our cohort nearly 57% of all coronary events in men occurred in subjects with total cholesterol <6.47 mmol/liter. … 62% [of the men who died of CHD in the MRFIT study (see Figure 3 subsequently)] had total cholesterol levels <6.22 mmol/liter (240 mg/dL) (38), and 38% had total cholesterol <5.7 mmol/liter (220 mg/dL)” (37, p. 681). The study of Livshits et al. also found “HDL-cholesterol percentages as a coronary risk indicator significantly surpasses the predictive value of total cholesterol” (37, p. 681).
- A World Health Organization expert committee published a report that said, “The majority of heart attacks occur in individuals with serum cholesterol levels below 240 mg/dL” (39).
If blood cholesterol concentrations are indeed lowered by Keys’ low-fat, low-cholesterol diet, what will be the benefits?
Using Framingham data available in 1975, H. M. Whyte (40) from the Australian National University in Canberra calculated that if 100 men with no other coronary risk factors were to lower their blood cholesterol concentrations from 310 to 260 mg/dL (8.0 to 6.7 mmol/L) for 20 years, starting at age 35, 94 would achieve no benefit whatsoever, whereas six could potentially benefit by avoiding a coronary incident. Of those 94 who would receive no benefit, eight would still suffer a heart attack even though they adhered strictly to the diet.
For 100 healthy men who lowered their blood cholesterol concentrations by the same amount but began only after they had reached age 55, only three would benefit. Benefits were somewhat greater for persons with multiple “risk factors” who were able to achieve greater reductions in their blood cholesterol concentrations.
Other researchers also developed models to predict what effect reducing the intake of dietary saturated fats, as required by Keys’ twin hypotheses, would have on life expectancy. W. C. Taylor et al. (41) predicted that if men at high risk of CHD because they smoked and had high blood cholesterol concentrations and blood pressures (and presumably insulin resistance) reduced their saturated fat intake, they might expect to live between 18 days and 12 months longer. But persons at low risk could expect “a gain in life expectancy of 3 days to 3 months from a lifelong program of cholesterol reduction” (p. 605).
Similarly, W. S. Browner and colleagues from the University of California, San Francisco (42), found, “A man who might otherwise die at sixty-five could expect to live an extra month if he avoided saturated fat for his entire life. If he lived to be ninety, he could expect an extra four months (of life)” (19, p. 65). S. A. Grover et al. (43) also calculated that men and women who reduced their dietary fat intakes in accord with national guidelines would reduce their blood cholesterol concentrations by a maximum of 29.1 mg/dL (0.75 mmol/L) and 21.4 mg/dL (0.55 mmol/L), respectively, and could expect an increase in life expectancy “by 0.03 to 0.4 year and 0.01 and 0.16 year respectively” (p. 1697). Smoking cessation, on the other hand, would increase life expectancies by between two and four years.
These data clearly show that efforts to reduce population smoking rates will have much greater impact on longevity than efforts to lower blood cholesterol concentrations by reducing dietary fat intake.
The first hint in the fhs that insulin resistance is a key determinant of CHD risk
So if blood total cholesterol concentration was not the key metabolic driver of CHD in the Framingham population, what was? A key finding was that HDL cholesterol appeared to be a protective factor against CHD (44). Castelli and Anderson (34) subsequently explained how they interpreted this finding:
Actually, there is probably a “symptom complex” in both sexes involving triglycerides that is strongly related to coronary heart disease. It is characterized by a normal level of cholesterol, a high level of triglycerides, and a low level of high-density lipoprotein (see Figure 2 subsequently) … Not only do persons with this complex have a higher incidence of coronary heart disease, they also have an incidence of diabetes mellitus that is twofold the rate in the general population and are usually overweight. Whether a high triglyceride level or obesity are truly independent risk factors in multivariate analysis in this setting may be academic. Clinicians should be aware of the constellation of findings: overweight, high triglycerides, average cholesterol, and low high-density lipoproteins all most likely adding to the risk of diabetes and coronary heart disease development. (34, p. 26, my emphasis and additions)
Readers of these columns will immediately realize these are all features of insulin resistance and consequent to eating high-carbohydrate diets by those who are insulin resistant (Figure 2).
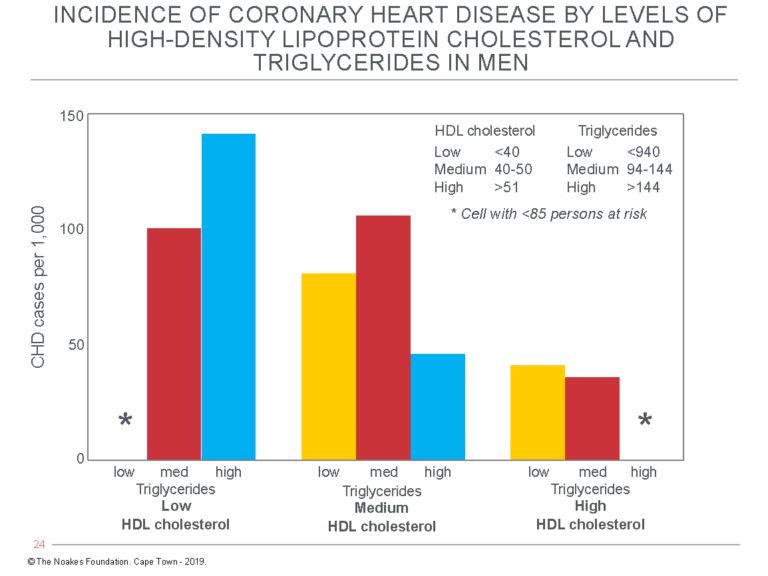
Figure 2: The publications of Castelli et al. (34) and Gordon et al. (44) included this figure to explain the relationship of the “symptom complex” they describe to CHD risk. The symptom complex they happened upon includes most of the features of the syndrome of insulin resistance. The graph shows the CHD risk (cases/1,000) as a function of three different blood triglyceride concentrations (low, medium, high) in persons with low, medium, or high blood HDL-cholesterol concentrations. Reproduced from references 34 and 44.
Figure 2 shows the lowest rates of CHD occurred in those male FHS subjects with the highest HDL cholesterol and medium blood triglyceride concentrations. The findings among female FHS subjects were identical (data not shown) (34, 44).
According to the theory that insulin resistance drives CHD rates, the lowest CHD rates should occur in those with the highest HDL cholesterol and lowest blood triglyceride concentrations (empty column on the extreme right), as this group would be the most insulin sensitive. But there are too few subjects in this category to calculate an accurate CHD risk. This means that few men (or women) in Framingham in the early 1960s were both insulin-sensitive and eating low-carbohydrate diets.
In contrast, the group at greatest risk comprises men with high blood triglyceride and low blood HDL-cholesterol concentrations (third column from left, counting also the extreme left column, which is empty). This group is, by our definition, the most insulin resistant. Therefore, according to the theory that insulin resistance is the true determinant of CHD risk, this group should be at the greatest risk for developing CHD, as is indeed the case. Figure 2 suggests those who are insulin resistant and eating high-carbohydrate diets are at three- to four-times greater risk for CHD than those who are the most insulin sensitive. This represents a difference of about 10 more deaths per annum for every 100 persons with those risky blood lipid profiles.
Compare this to the far more modest, essentially irrelevant increases in risk predicted by rising blood cholesterol concentrations (Figure 1).
Note, also, that the column on the extreme left is likewise empty. This is because there will always be few persons with simultaneously low HDL cholesterol and low blood triglyceride concentrations.
There is presently no known dietary combination that produces this outcome.
MRFIT data confirm blood cholesterol concentration is a weak predictor of CHD risk
For completeness, it’s relevant to include one other graph from the same book (32). Figure 3 shows the same relationship between blood cholesterol concentrations and annual death rate from data collected by Keys’ two greatest SCS buddies, Drs. Kannel (45) and J. Stamler (38), in the Multiple Risk Factor Intervention Trial (MRFIT) involving more than 362,000 men (46).
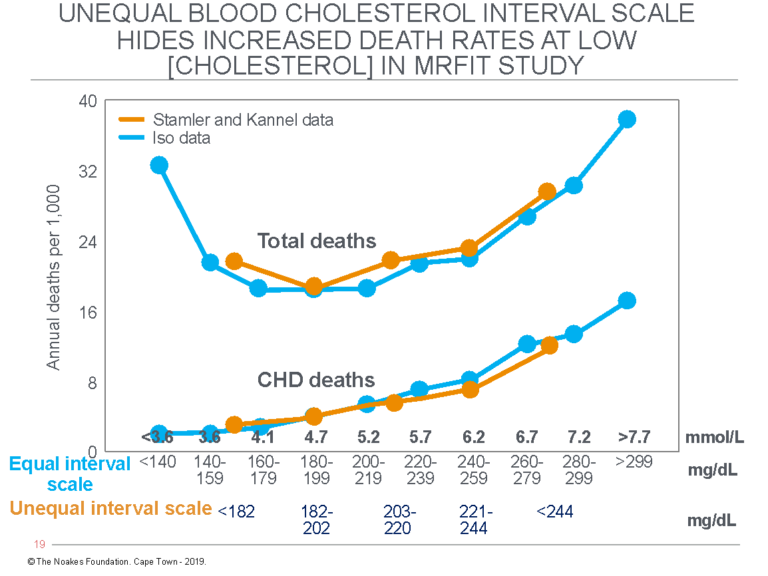
Figure 3: The figure shows the annual CHD and total death rates per 1,000 individuals among participants in the MRFIT with different blood cholesterol concentrations. The data of Kannel et al. (45) and Stamler et al. (38) (orange lines) are divided into groups that have unequal intervals for blood cholesterol concentrations. In particular, the group with the lowest blood cholesterol concentrations includes a much wider range of concentrations. The result, which appears willful on the part of the researchers (32), is to hide an increase in total deaths at low blood cholesterol concentrations. This is exposed by the manner in which H. Iso et al. (47) presented the same data (blue lines).
Figure 3 compares the annual CHD and total death rates for different blood cholesterol concentrations as presented by different authors using the same data. The data of Kannel et al. (45) and Stamler et al. (38) (orange line) are presented with an unequal cholesterol interval scale that is truncated both in the bottom and top divisions. Thus, the lowest interval includes blood cholesterol concentrations from <140 mg/dL to 182 mg/dL, and the highest, from 244 mg/dL to >299 mg/dL. The other three categories include a much narrower range of blood cholesterol concentrations of only ~20 mg/dL.
Iso et al. (47) presented exactly the same data but divided into 10 groups; eight with equal (19 mg/dL) ranges of blood cholesterol concentrations. Only the bottom and top categories include a wider range of blood cholesterol concentrations.
By removing the truncation at the bottom end of the curve, the data of Iso et al. (47) clearly established that, whereas total CHD deaths are slightly reduced at the very lowest blood cholesterol concentrations, total deaths increase at those blood cholesterol concentrations. Predictably, this had to be hidden by Keys’ buddies who were promoting the lipid hypothesis, because that hypothesis predicts that the lower the blood cholesterol concentration, the more benefit the patient will receive. Indeed, this is what most lay people have been taught over the years.
Instead, what these data show is that low blood cholesterol concentrations are associated with increased deaths from non-cardiac causes — exactly what was found in the SCS (10).
Of even greater interest is the magnitude of the effect of an increasing blood cholesterol concentration on risk of CHD death. The graph shows an increase in blood cholesterol concentration from <140 mg/dL (3.6 mmol/L) to >299 mg/dL (7.7 mmol/L) increases the annual risk of CHD death from about 0.4/1,000 (0.04%) to about 2.7/1,000 (0.27%). Thus, for the individual, the risk rises by 0.23 percent per annum for a 213% increase in blood cholesterol concentration.
This means 97.7 of 100 middle-aged persons with blood cholesterol concentrations “dangerously elevated” at >299 mg/dL (>7.7 mmol/L) will still be alive 10 years after being told they are at extreme risk of dropping dead the next day from an imminent heart attack. This applies even to those who do absolutely nothing to improve their health during that 10 years.
But in the range of more usual blood cholesterol concentrations between 180 mg/dL (4.7 mmol/L) and 220 mg/dL (5.7 mmol/L), the increase in risk is from 0.5/1,000 (0.05%) to 1.0/1,000 (0.10%), or about 0.05% per annum.
Thus, Figure 3, like Figure 1, establishes once and for all that blood cholesterol concentration is a very poor predictor of future CHD risk. Certainly, it is not sufficiently powerful to mandate a global change in eating behaviors, as happened when the 1977 U.S. Dietary Guidelines were promulgated (48). Nor could it ever possibly justify the use of powerful and potentially toxic cholesterol-lowering drugs to reduce blood cholesterol concentrations to the “normal” range.
Looking at the data in Figures 1 and 3, it is rather difficult to decide exactly what should be the “normal” range for blood cholesterol concentrations.
The sole conclusion one might draw from Figures 1 and 3 is that an elevated blood cholesterol concentration cannot be the key driver of coronary atherosclerosis and CHD. Something else must be much more important.
Smith and Pinckney (32) first highlighted these findings in 1991, and Gary Taubes brought them to the attention of a wider audience through a detailed analysis in his landmark 2007 book, Good Calories, Bad Calories (19, pp. 63-65). Nevertheless, this information, which essentially destroys Keys’ lipid hypothesis, has yet to pierce the consciousness of either the medical profession or general public. One might ask: How much longer will it be suppressed?
With time, the researchers in the FHS seemingly saw this truth, since they became much less interested in blood cholesterol concentrations as the key risk factor for CHD. Rather, the FHS acquired a new role: “epidemiological activism” promoting the relationship of elevated blood pressure (hypertension) to increased risk of stroke and congestive heart failure (6), a relationship for which there is much irrefutable scientific evidence.
But for some reason, the hypertension/stroke/heart failure link has never quite enjoyed the same intellectually seductive attraction as the diet-heart and lipid hypotheses.
More commercial forces driving Keys’ lipid hypothesis
In 1985, Drs. Joseph Goldstein and Michael Brown were awarded the Nobel Prize in Medicine (49) for their discovery that LDL cholesterol enters cells via a specific LDL receptor and that this process is defective in persons with very high blood cholesterol concentrations on a genetic basis — so-called familial hypercholesterolemia (FH) (50, 51). They next discovered why this inability of persons with FH to import LDL cholesterol into their cells explains their very high blood cholesterol concentrations. For it is the cholesterol imported into the cell via this mechanism that regulates how much cholesterol each cell will produce. In the liver cells, this process regulates how much cholesterol the liver will produce to maintain a particular blood cholesterol concentration.
Fatefully for all of us, Goldstein and Brown also determined the mechanism by which the imported cholesterol regulates cholesterol production. It does so by controlling the rate of action of the enzyme 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMG CoA reductase), which is the key enzyme regulating the synthesis of cholesterol and consequently a number of other chemicals (the steroid hormones: glucocorticoids, mineralocorticoids, androgens, estrogens, and progesterones; the bile acids; and vitamin D), all of which are derived from the cholesterol produced in this metabolic pathway.
Since Goldstein and Brown knew persons with FH have a somewhat higher risk for developing CHD — that, after all, was why they chose this particular research direction — they naturally understood that they had uncovered a potential goldmine for the pharmaceutical industry. What was now required was the discovery of a compound that could block HMG CoA reductase activity. As they wrote, “The potential clinical application of these new data is being explored further. Hopefully it may one day be possible to develop a specific therapy for familial hypercholesterolemia” (51, p. 150).
This is a very interesting statement given that the prevalence in the population of FH ranges from 1 in 500 (heterozygous FH) to 1 in 106 (homozygous FH). Clearly Goldstein and Brown did not originally consider that their finding should be applied to lower the blood cholesterol concentrations of the entire world.
It would not take long, in fact just 17 months, for Goldstein and Brown to discover that a Japanese investigator, Akira Endo, had isolated a molecule he called ML-236B (compactin) from a fungal (penicillin) mold. Compactin turned out to be a potent competitive inhibitor of HMG CoA reductase (52). In time, their collaborations led to the commercial production of the cholesterol-lowering drugs, the statins.
During this collaboration, Goldstein and Brown (53) became interested in the possibility that the HMG CoA reductase inhibitors might also act by increasing the number of LDL receptors on cells, especially liver cells. They recruited Dr. Scott Grundy to their laboratories to be a key player in this part of their research.
Within a short time, the team had produced evidence that the HMG CoA reductase inhibitor, Mevinolin, does indeed have this effect (54).
Grundy had, perhaps unwittingly, become another important member of the team promoting Keys’ lipid hypothesis.
Grundy helps spin the MRFIT cholesterol/CHD relationship as the greatest financial scam in medical history is about to take off
Galvanized by his discovery that HMG CoA reductase inhibitors could stimulate the production of liver cell LDL receptors and might provide a pharmacological solution to the global heart disease epidemic, Grundy (36) wrote a State of the Art/Review in the influential Journal of the American Medical Association in 1986. He began by noting Goldstein and Brown had just been awarded the 1985 Nobel Prize. Next, citing his own work (54), he reported that a new class of drugs, the HMG CoA reductase inhibitors, with potent cholesterol-lowering potential, had recently been developed. This was naturally of great interest to the medical profession since the Lipid Research Clinics (LRC) Coronary Primary Prevention trial had established that drug-induced lowering of blood cholesterol concentrations “reduces the frequency of several manifestations of CHD, including myocardial infarction” (36, p. 2849).
In reality, as Taubes has described, the $150-million LRC Coronary Prevention Trial was a complete and utter failure (19, pp. 56-58). The key outcome was that whereas 71 men in the control group died during the trial, 68 died in the intervention group, for a difference of 0.2% (54, 55). On the basis of this finding, the senior author of the trial, Basil Rifkind, MD, told Time magazine, “It is now indisputable that lowering cholesterol with diet and drugs can actually cut the risk of developing heart disease and having a heart attack” (19, p. 57). He later explained to Taubes that he had willfully misrepresented the negative findings of the LRC trial (19, p. 58): “‘It’s an imperfect world,’ Rifkind said. ‘The data that would be definitive is ungettable, so you do the best with what is available’” (p. 58) — even if you are reduced to lying, it seems.
In his introduction, Grundy indicated he too was wedded to Keys’ lipid hypothesis. Predictably, he would interpret any and all data as favorable to that hypothesis. Given his links to Goldstein and Brown, and through them to the imminent commercialization of the blockbuster statin drugs, how could he possibly hold any other beliefs? After all, the laboratory had secured a Nobel Prize. What chance of a second?
In the article, Grundy reproduced the MRFIT data from Figure 3 with the addition of a graph showing the risk ratio for CHD mortality at different blood cholesterol concentrations (Figure 4).
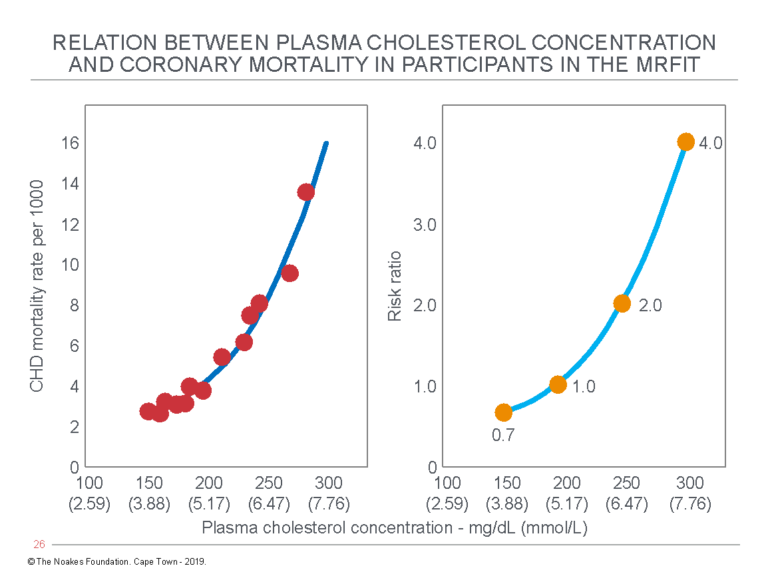
Figure 4: In his 1986 State of the Art/Review, Grundy (36) reproduced the MRFIT data, also used to construct Figure 3 (left panel). In the right panel, he showed the risk ratio for CHD mortality apparently rises from 0.7 for those with blood cholesterol concentrations of 150 mg/dL (3.9 mmol/L) to 4.0 for those with blood cholesterol concentrations “elevated” to 300 mg/dL (7.8 mmol/L). Naturally, these are the figures that are taught to medical students during their training. They will predictably generate a natural desire to lower blood cholesterol concentrations to “as low as possible” to reduce CHD risk to “as low as possible.” But the truth is rather more nuanced. Reproduced from reference 36.
By adding the right panel in Figure 4, Grundy upped the stakes by suggesting a high blood cholesterol concentration of 300 mg/dL (7.8 mmol/L) raises the risk of death from CHD four-fold. But as I have argued earlier in relation to Figure 3, if the risk associated with low blood cholesterol concentration is very low, then raising that very low risk four-fold still makes the risk very low.
Grundy then introduced another figure (Figure 5), based purely on speculation, which tried to explain these mathematical findings in terms of what is happening to the coronary arteries of persons with different blood cholesterol concentrations. Importantly, this graph is not based on definitive evidence; it is based purely on hypothetical projection. But when reproduced in an influential medical journal read by tens of thousands of doctors and scientists and coming from the laboratory of two Nobel Prize winners, it will generate a truth and life all its own.
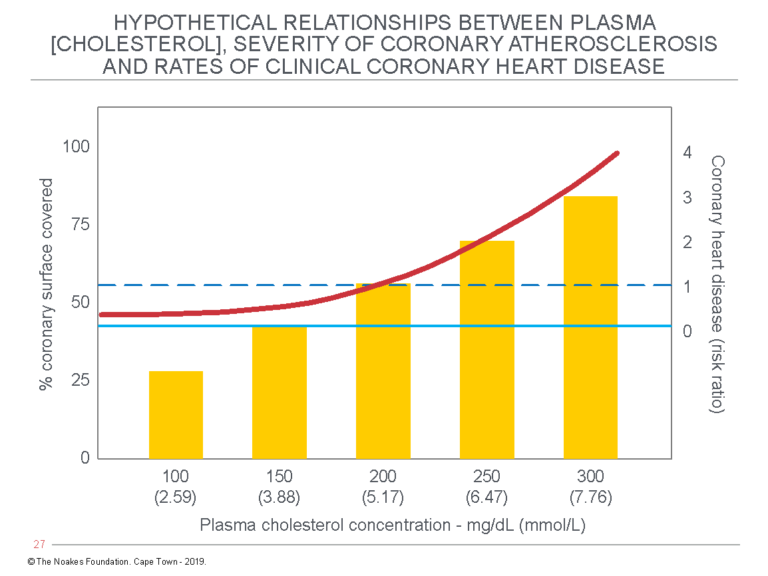
Figure 5: Grundy’s article includes this figure, which describes the “hypothetical relationships among plasma cholesterol levels, severity of coronary atherosclerosis, and rates of clinical coronary heart disease.” The emphasis should be that the figure is indeed “hypothetical” and utterly dependent on the lipid hypothesis being completely correct. Reproduced from reference 36.
The most important point about Figure 5 is that it is entirely hypothetical. In essence, it proposes that the extent of coronary artery atherosclerosis (the % coronary surface covered by atherosclerosis) is a curvilinear function of the blood cholesterol concentration. According to Grundy, this then is the sole explanation for why there is a curvilinear increase in CHD risk ratio associated with increasing blood cholesterol concentrations.
Recall that association does not prove causation, and here, Grundy is using associational ecologic data as proof of unseen and unmeasured pathological processes in human coronary arteries. This is not good science.
Perhaps we should call this the Grundy amplification of Keys’ lipid hypothesis. It is no more credible.
In the first place, the level of blood cholesterol concentration has never been shown to be a predictor of the extent of coronary atherosclerosis (57). Instead, W. R. Ware cites 19 studies that have shown there to be no such relationship.
In the second place, heart attack and death from CHD involve at least three processes that are likely unrelated to specific risk factors:
- Coronary atherosclerosis
- The development of instability of the coronary plaque leading to plaque rupture
- Arterial thrombosis (hence the original term for heart attack was a coronary thrombosis)
The problem with Figure 5 is that the “coronary disease risk ratio” is the result of at least three separate biological processes, only one of which is coronary atherosclerosis. The figure then hypothesizes that the contribution of all three can be predicted by a single blood marker: the blood cholesterol concentration. We now know this is simply false.
And in the third place, so what if the blood cholesterol concentration does indeed predict all these outcomes? The left panel of Figure 4 (like Figure 3) predicts an increase in blood cholesterol concentrations from 150 mg/dL (3.88 mmol/L) to 300 mg/dL (7.76 mmol/L) increases annual mortality from 3/1000 to a staggering 16/1000, or from 0.3% to 1.6%, an absolute increase of 1.3%. This is not the 400% increase Grundy wished to convey (Figure 4, right panel).
So all that Grundy’s figures proved, once more, was that if the blood cholesterol concentration does indeed play some role in the development of CHD, its role is so small as to be almost immeasurable.
Unfortunately, Grundy’s State of the Art/Review became part of the global marketing of the new group of drugs, the statins, which would be prescribed to tens of millions over the next three decades and generate trillions of dollars in income.
But anyone who now understands the real meaning of Figures 3 and 4 should realize the ultimate goal — trying to beat a disease to which the blood cholesterol concentration makes no meaningful contribution by lowering the blood cholesterol concentration — would not produce a positive outcome.
The Framingham study confirms the importance of insulin resistance as a risk factor for the future development of coronary heart disease
Ironically, what the original design of the Framingham study failed to achieve may have been resolved by the follow-up study, which investigated the future health of the children of the original FHS cohort. So a 14-year follow-up study of 2,910 of the offspring of the original FHS cohort found the key predictor of subsequent mortality was the presence or absence of insulin resistance (58) and not blood cholesterol concentration (Figure 6).
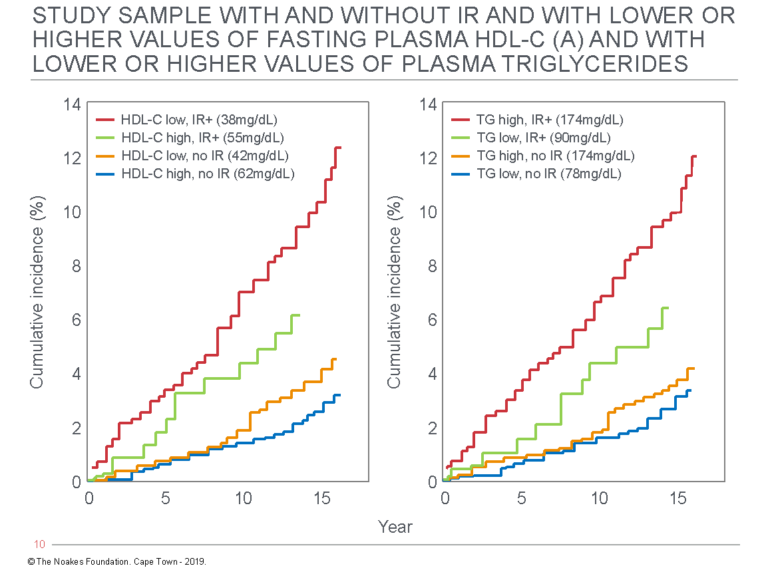
Figure 6: This figure shows the cumulative incidence of new CHD events in the Framingham Offspring Study based on the presence or absence of insulin resistance with higher or lower blood HDL-cholesterol concentrations (left panel) and the presence or absence of insulin resistance with either higher or lower blood triglyceride concentrations (right panel). Compared to those with low HDL cholesterol or high triglyceride levels and insulin resistance, the cumulative incidence of events is three times lower in those with similarly abnormal blood HDL cholesterol or triglyceride values if they are insulin sensitive. Reproduced from reference 58.
Thus persons with low blood HDL cholesterol and high blood triglyceride concentrations were at three times greater risk for developing a CHD event only if they were also diagnosed as insulin resistant by the HOMA-IR test. Conversely, having higher HDL-cholesterol and lower triglyceride concentrations exactly halved risk of future events in those with insulin resistance.
Naturally, those who were insulin sensitive with high HDL cholesterol and low triglyceride concentrations were at the lowest risk for future events. In fact, their future event rates were about one-sixth those experienced by persons who were insulin resistant but who also had low HDL cholesterol and high triglyceride concentrations.
All these findings are predictable on the basis of the findings in the Framingham parents (Figure 2; 34, 44).
Two final questions
1. What if risk factors are not risk factors after all?
As I worked through the material for these columns, the realization finally struck me (after 44 years as an MD) that medical science has failed to prove that the concept of CHD risk factors is a viable concept with major practical value (other than to produce a financial return for some).
For example, we are forever told that the FHS was established to identify metabolic and other risk factors for the future development of CHD. What the FHS clearly established is that blood cholesterol concentration has no value in predicting such risk (Figure 1), a finding confirmed by the MRFIT trial (Figures 3 and 4). The finding that insulin resistance is the key driver for CHD even in the Framingham data (Figures 2 and 6) is not something that will ever be widely communicated by the medical and scientific communities. The truly important finding of the FHS simply will remain buried.
My conclusion is that there is good evidence for four factors that definitely modify the risk for developing CHD: smoking, hypertension, insulin resistance/Type 2 diabetes mellitus, and physical activity. There are strong and established biological reasons to explain why these four factors are important. But for the rest — especially the lipid panels beloved of cardiologists — well, to me, it just seems to be guesswork, premised on Keys’ disproven hypotheses.
In my opinion, and based on my understanding of the current evidence, the most important effects of what we choose to eat from the foods currently available to us are determined by whether those food choices either lessen or worsen each individual’s degree of insulin resistance, hyperinsulinemia, hyperglycemia, and hypertriglyceridemia; or cause or prevent non-alcoholic fatty liver disease (NAFLD).
This belief is strengthened by the finding that the MRFIT trial (Figure 3), which studied 12,866 men, 6,428 of whom were subjected to “an in-depth sustained multifactor intervention program aimed at lowering serum cholesterol and BP and smoking cessation,” failed to produce a reduction in the primary endpoint, fatal CHD (59). A subsequent post-hoc analysis published 27 years later (60) found a small, 1.3% reduction in combined fatal and non-fatal events, which was a significant outcome in favor of the intervention group.
But to achieve this statistically significant difference would require that 77 persons would need to follow the intervention religiously for seven years for one of them to benefit. We also do not know if any of the 76 who did not benefit from the intervention might actually have suffered some unexpected harm.
I think it is fair to conclude interventions based on attempts to reverse what we believe to be key CHD risk factors have not produced the outcomes we expect based on the premise that these surrogate markers really do act as risk factors for clinical disease. This must suggest that our basic understanding of the predictive value of CHD risk factors could well be wrong.
My concerns were strengthened by reading the paper by T. E. Strandberg et al. (61), which, like the MRFIT study, reported the effects of an intensive intervention program, this time in 1,222 middle-aged men in Helsinki. The study was randomized and controlled and lasted five years, during which the 612 men in the intervention group visited the investigators every four months. Those with hypertension or raised blood cholesterol concentrations were treated with the “appropriate” drugs.
The members of the intervention group were also advised to stop smoking, achieve a normal body weight through the adoption of a prescribed diet, and moderate their alcohol intake.
The 610 men in the control group received no such interventions. Both groups were examined clinically after two and then every five years. The intervention group achieved a significant 46% reduction in CHD risk factors. By five years, about half of the men in the intervention arm were receiving at least one medication to manage their risk factors.
During the first 15 years of the trial, there were 67 deaths in the intervention group and 46 in the control group. Excess deaths in the intervention group were caused by CHD (34 vs. 14 deaths) and violence (13 vs. 1). Deaths from cancer were not different between groups.
Figure 7 shows that deaths from all causes accumulated more rapidly in the intervention group than the control group. The same applied to deaths assigned to heart disease. In other words, the intervention substantially worsened outcomes.
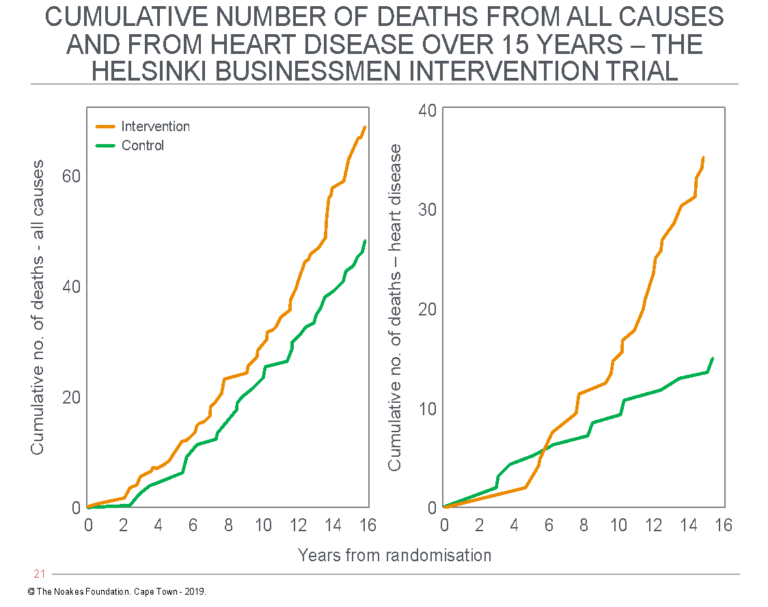
Figure 7: The figure shows the cumulative incidence of deaths from all causes (left panel) and deaths assigned to cardiac disease (right panel) in the study of Strandberg et al. (61), the Helsinki Businessmen Study. Data for the intervention group are represented with the orange line; the control (non-intervention) group with the green line. Note that deaths are increased in the intervention group. Reproduced from reference 61.
So the key conclusion from the study is that the intervention, based on the hypothesis that the reversal of coronary risk factors must reduce deaths from CHD and, as a result, also from all causes, produced the opposite result. The intervention caused harm.
The authors could only suggest that “these unexpected results may not question multifactorial prevention as such” (61, p. 1225) — except, of course, in the process of normal science, these findings should most definitely raise questions about whether we should be forcing well-intended interventions on patients to reverse their “risk factors” when the outcome may be harmful to their health.
Recall what was done to President Dwight D. Eisenhower (62) under the guise of novel ground-breaking, scientific thinking (hypotheses). But which ideas have turned out to be false?
2. Are heart attacks and strokes really the result of identical biological mechanisms?
I was introduced to another paradox highlighted by Terence Kealey (63). He has drawn attention to this paradox that has been hidden in the diet-heart debate. It is the general presumption, stemming directly from Keys’ hypotheses and still taught in the medical and dietetics professions, that atherosclerosis, caused by identical biological mechanisms, causes strokes and heart attacks. The sole difference is that the disease occurs at different anatomical sites: in the cerebral arteries causing strokes and in the coronary arteries causing heart attacks.
Logically, if a diet-induced increase in blood cholesterol concentrations is the cause of the rising CHD from 1920 to 1950, then exactly the same pattern must have happened for the incidence of stroke. But Figure 8 shows this is not the case.
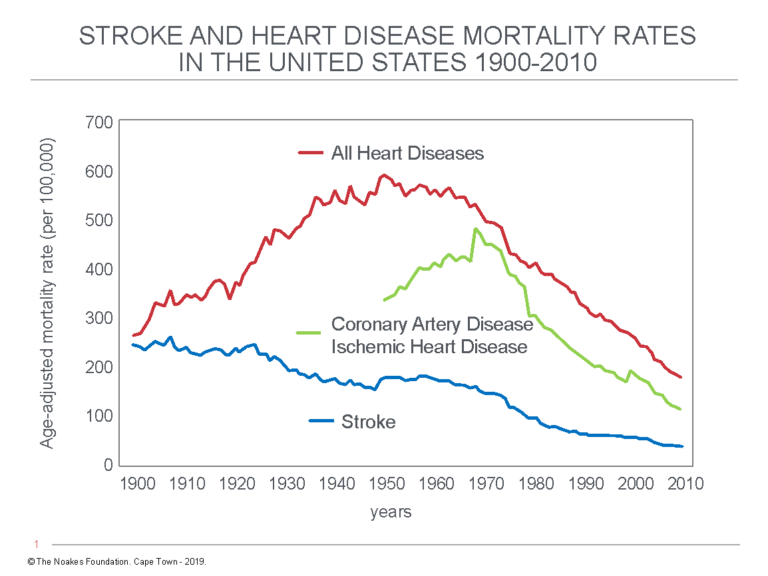
Figure 8: The graph for the incidence of all heart diseases (red line) mirrors the rise and fall of coronary artery disease (CHD) and ischemic heart disease (IHD) incidence rates (green line) from 1900 to 2010. This confirms coronary atherosclerosis causing CHD/IHD made the major contribution to changing heart disease death rates during this time period. Stroke rates (blue line) have fallen progressively since 1900. This is paradoxical. Redrawn from reference 64.
Figure 8 shows the incidence rates for strokes and coronary heart disease/ischemic heart disease (CHD/IHD) have followed different trajectories since 1900 (64). Whereas CHD/IHD rates have fallen continuously since 1900, CHD/IHD death rates have increased dramatically before decreasing just as dramatically.
It is difficult to believe that this difference can be explained by a common mechanism: a diet-induced increase in blood cholesterol concentrations increasing mortality from CHD/IHD but at the same protecting the cerebral arteries from the development of atherosclerosis causing strokes.
I wonder if Keys ever considered this paradox or how he would explain it — or indeed what anyone who prescribes cholesterol-lowering drugs and a low-fat diet for persons who have cerebrovascular disease would say.
So what outcomes did the FHS produce?
Although Keys was not directly involved in the FHS, in a sense it was his drive to find funding for his Minnesota Business and Professional Men Study (5) that brought the FHS into existence. Paul Dudley White was also involved. It was on White’s advice that the study was located in Framingham, conveniently, in close proximity to the many eager cardiologists at Harvard Medical School (6, p. 1001).
The goal of the FHS was to determine whether it is possible to predict the future development of CHD through the measurement of what became known as “risk factors” in persons initially free of CHD. Risk factor assessment is now an established feature of clinical medicine and explains why doctors measure and then treat prospectively, for example, elevated blood pressure readings and “high” blood cholesterol concentrations in selected patients. The assumption is that such treatment prevents or reduces the manifestations of CHD. The outcomes in Figure 7 indicate that we should not accept that prediction uncritically.
The question needs to be asked: What did the FHS actually deliver in terms of identifying CHD risk factors?
First, the study was unable to provide any evidence that a high-fat diet caused elevated blood cholesterol concentrations (9, 14, 19, 20, 21). Thus, the FHS disproved Keys’ diet hypothesis, which, recall from an earlier column (12), was also originally based on associational studies. These were of the same kind as the FHS but with much less robust dietary analyses (9, 13). Thus, any reasonably independent scientists should have declared that the FHS buried Keys’ diet-heart hypothesis.
Instead, Kannel, in his position as director of the FHS, simply obfuscated with circumlocution by saying, “Although there is no discernible relationship between reported diet intake and serum cholesterol levels in the Framingham Diet Study group, it is incorrect to interpret this finding to mean that diet has no connection with blood cholesterol … since the evidence incriminating diet in producing elevated serum cholesterol concentrations is quite substantial” (9, p. 7).
Once more Kannel is guilty of misunderstanding how science should be conducted (17). If one sets up a hypothesis and then collects evidence that inconveniently disproves one’s hypothesis, then it matters not how beguiled one is with that hypothesis. It must be rejected and replaced with a fresh hypothesis ready to be tested. In this way, science advances from one failed and disproven hypothesis to the next.
But scientists corrupt the scientific process when they refuse to acknowledge their data disprove their beloved hypotheses and instead use ad-hoc excuses to explain why the data are wrong and hence cannot be trusted. This, precisely, is the method used by Kannel.
The problem with this model of scientific endeavor is that the only way for science to advance toward the truth is for the perpetrators of the fake science to die. As Nobel laureate German theoretical physicist Max Planck famously remarked, “A new scientific truth does not triumph by convincing its opponents and making them see the light, but rather because its opponents eventually die, and a new generation grows up that is familiar with it.” Economist Paul A. Samuelson shortened this idea: “[This type of] Science advances funeral by funeral.”
Through his use of this scientific deception, Kannel joined the senior scientists conducting the $700-million Woman’s Health Initiative, which evaluated the effects of a Keysian low-fat, low-cholesterol diet on the future health of post-menopausal women (65). When they discovered the Keys-inspired diet they tested failed to improve health and may even have been detrimental for many, they hid the facts and simply blamed the subjects for not trying hard enough (66; 67 p. 61-62). Naturally, the hypothesis of the “nutrition aristocrats” (14, p. 69) could never be wrong.
Kannel and his colleagues would do well to remember the quote of intellectual giant Richard P. Feynman: “It doesn’t matter how beautiful your theory is, it doesn’t matter how smart you are. If it doesn’t agree with experiment, it’s wrong.”
Mann, one of the founding scientists behind the FHS but who resigned when he lost faith in the integrity of those involved in the study, explained why he thinks this happens to so many scientists:
Why do scientists do this? There are, I think, three reasons. They seem not to understand the way science operates. We advance in science by disproving a hypothesis. When one is disproven it is replaced with a new hypothesis and a new trial is mounted to see if that can be disproved. These people are arrogantly prideful, full of hubris. They are unable to discard an untrue proposition because they see it as their creation. They seem unable to admit error, which is the very substance of progress in science … (68, p. 215)
The second finding of the FHS was the inability of the blood cholesterol concentration to make any reasonably practical contribution to the prediction of future risk for CHD (Figure 1; 26, 28-30, 33-35). In the range of blood cholesterol concentrations between 140 to 259 mg/dL (3.6-6.7 mmol/L), total annual death rates were very low and did not change (Figure 1).
If ever there was proof that the blood cholesterol concentration is not the key driver of CHD as Keys’ lipid hypothesis predicts, then Figures 1, 3, and 4 are the best evidence.
Thus, properly interpreted, the FHS should have buried Keys’ lipid hypothesis — or, it should at least have forced the realization that even if the blood cholesterol concentration perhaps contributes something to CHD risk, its contribution is probably trivial.
The one crowning achievement of the FHS was to show very early on that elevated blood pressure increases risk of CHD four-fold (26) and also increases risk of stroke (69). Interestingly, risk of stroke was unrelated to the blood cholesterol concentration (69), which should have been of some interest to Keys and his acolytes (Figure 8). Subsequent studies have confirmed that hypertension is a significant risk factor for both stroke (70) and heart failure (71, 72).
The FHS clearly established elevated blood pressure is an important risk factor for CHD, heart failure, and stroke. But it failed miserably to support Keys’ twin hypotheses. This point is not properly emphasized in medical education in my opinion.
Dr. Walter Willett is one of the most published scientists in the field of nutrition. A vegetarian in his own life, Willett is most certainly not a fan of either a meat-based or low-carbohydrate diet. In fact, he is quite a fan of Keys’ hypotheses (73).
But in 1990, he summarized what he believed had been achieved by the nutritional research completed up to that time (74). This would have included all of Keys’ research and that of the FHS and MRFIT trials, among many others:
Despite the prominence given to the relation between diet and coronary heart disease (CHD) during the last 40 years, direct evidence is disappointingly meagre. Very few human studies, either case-control or cohort observational studies or intervention trials, have directly examined the relationships between dietary factors and cardiovascular disease. Almost all were seriously limited in size or by their method of dietary assessments (75). Only two consistent findings have emerged:
• an inverse relation between energy intake and the risk of coronary heart disease (almost surely reflecting the protective effects of physical activity), and
• a strong protective effect of moderate alcohol intake.
Although the focus of dietary recommendations is usually a reduction in saturated fat intake, no relation between saturated fat intake and risk of CHD was observed in the most informative prospective study to date (76). Even if such a relationship exists, the widely quoted ecological association found by Keys (77) was probably fortuitously confounded, because the magnitude of the relationship between saturated fat intake and CHD rates was considerably stronger than expected on the basis of metabolic studies of diet and blood lipids and, in turn, epidemiologic studies of serum lipids and CHD risk (76).
The classical diet/heart hypothesis — that risk of CHD is increased by dietary cholesterol and saturated fat and reduced by polyunsaturated fat — probably contains some element of truth. However, the hypothesis is over-simplified in many respects and needs continued refinement. For example, it has been known for decades, but largely ignored, that only some saturated fats increase serum cholesterol … . Thus, as pointed out by Grundy (78), the use of the generic term “saturated fat” to describe diets may be outmoded, both in research, and in policy and consumer education. (p. 1295-1296)
Willett added: “As the scientific basis for a reduction in total fat is weak, and the concept of generic ‘saturated fat’ is increasingly outmoded, nutrition educators and those involved in food labelling are challenged to provide clear and scientifically sound messages for the public” (p. 1297).
This would seem to be a fair review of the extent to which Keys’ work has failed to provide definitive evidence linking a particular diet with the causation of CHD.
But the most definitive statement on the failure of Keys’ hypotheses belongs to the man who was initially involved with the FHS but then deserted what he perceived to be the “greatest health scam of the century” (13, p. 1).
Mann was relentless in his criticism of the hapless contributions of Keys and others:
Our disagreement with Keys and Stamler and their sycophants is in management. They claim that dietary manipulation of fat and cholesterol will reduce hypercholesterolemia and thereby diminish CHD. They have not shown this to be true, and, more importantly, they have not shown that any dietary or drug treatment of any combination of them is safe. It is time now for scientists and regulators to correct these grave errors, because these pseudo-cures prevent the development of safe and efficacious remedies. (79, p. 74)
Mann continued:
I started out in biomedical science because I felt science was the purest, cleanest, and most useful of all human endeavours. Science seemed above and beyond selfish scheming and falsehood. It was not free of error, but it was surely free of cheating because that would be exposed by measurement — by the truth. Now I am older and wiser, and I see that I was mistaken. We do have fraud and deception in science. We have painted mice, plagiarism, fraudulent psychometrics, stolen manuscripts, faked physiologic data, and cheating in molecular biology. But the most immense fraud of all is here in the very field where I have worked. (79, p. 74-75)
In the final column reviewing Keys’ legacy (3), I explain the steps he took to ensure his hypotheses would grow to dominate global teaching in both medicine and dietetics.
Finally, I provide a list of the intellectual and scientific crimes of which I consider him to be guilty.
ADDITIONAL READING
- It’s the Insulin Resistance, Stupid: Part 1
- It’s the Insulin Resistance, Stupid: Part 2
- It’s the Insulin Resistance, Stupid: Part 3
- It’s the Insulin Resistance, Stupid: Part 4
- It’s the Insulin Resistance, Stupid: Part 5
- It’s the Insulin Resistance, Stupid: Part 6
- It’s the Insulin Resistance, Stupid: Part 7
- It’s the Insulin Resistance, Stupid: Part 8

Professor T.D. Noakes (OMS, MBChB, MD, D.Sc., Ph.D.[hc], FACSM, [hon] FFSEM UK, [hon] FFSEM Ire) studied at the University of Cape Town (UCT), obtaining a MBChB degree and an MD and DSc (Med) in Exercise Science. He is now an Emeritus Professor at UCT, following his retirement from the Research Unit of Exercise Science and Sports Medicine. In 1995, he was a co-founder of the now-prestigious Sports Science Institute of South Africa (SSISA). He has been rated an A1 scientist by the National Research Foundation of SA (NRF) for a second five-year term. In 2008, he received the Order of Mapungubwe, Silver, from the President of South Africa for his “excellent contribution in the field of sports and the science of physical exercise.”
Noakes has published more than 750 scientific books and articles. He has been cited more than 16,000 times in scientific literature and has an H-index of 71. He has won numerous awards over the years and made himself available on many editorial boards. He has authored many books, including Lore of Running (4th Edition), considered to be the “bible” for runners; his autobiography, Challenging Beliefs: Memoirs of a Career; Waterlogged: The Serious Problem of Overhydration in Endurance Sports (in 2012); and The Real Meal Revolution (in 2013).
Following the publication of the best-selling The Real Meal Revolution, he founded The Noakes Foundation, the focus of which is to support high quality research of the low-carbohydrate, high-fat diet, especially for those with insulin resistance.
He is highly acclaimed in his field and, at age 67, still is physically active, taking part in races up to 21 km as well as regular CrossFit training.
References
- Anderson KM, Castelli WP, Levy D. Cholesterol and mortality. 30 years of follow-up from the Framingham Study. JAMA 257(1987): 2176-2180.
- Ramsden CE, Zamora D, Majchrzak-Hong S, et al. Re-evaluation of the traditional diet-heart hypothesis: analysis of recovered data from Minnesota Coronary Experiment (1968-73). BMJ 353(2016): i1246.
- Noakes TD. It’s the insulin resistance, stupid: Part 10. Forthcoming on CrossFit.com.
- Keys A. The inception and pilot surveys. In: Kromhout D, Menotti A, Blackburn H. The Seven Countries Study. A scientific adventure in cardiovascular disease epidemiology. Marjan Nijssen-Kramer. Bilthoven, Netherlands: Studio RIVM, 1993. pp.15-26.
- Keys A, Taylor HL, Blackburn H, et al. Coronary heart disease among Minnesota business and professional men followed fifteen years. Circulation 28(1962): 381-394.
- Mahmood SS, Levy D, Vasan RS, et al. The Framingham Heart Study and the epidemiology of cardiovascular disease: A historical perspective. Lancet 383(2014): 999-1008.
- Eades M. The Framingham Heart Study, Part 1: Cargo-cult science. CrossFit.com. 13 Jan. 2019. Available here.
- Eades M. The Framingham Heart Study, Part 2: The Framingham observation. CrossFit.com. 22 Jan. 2019. Available here.
- Eades M. The Framingham Heart Study, Part 3: Framingham’s presentational flaws – Bias or Fraud? CrossFit.com. 27 Jan. 2019. Available here.
- Noakes TD. It’s the insulin resistance, stupid: Part 7. CrossFit.com. 11 Nov. 2019. Available here.
- Jacobs DR, Anderson JT, Hannan P, et al. Variability in individual serum cholesterol response to change in diet. Arteriosclerosis 3(1983): 349-356.
- Noakes TD. It’s the insulin resistance, stupid: Part 6. CrossFit.com. 30 Oct. 2019. Available here.
- Mann GV. A short history of the diet/heart hypothesis. In: Coronary Heart Disease. The dietary sense and nonsense. An evaluation by scientists. Mann GV, Ed. London, England: Janus Publishing Company, 1993. pp.1-17.
- Teicholz N. The Big Fat Surprise. Why Butter, Meat and Cheese Belong in a Heathy Diet. New York, NY: Simon and Schuster, 2014.
- Jacobs DR, Anderson JT, Blackburn H. Diet and serum cholesterol. Do zero correlations negate the relationship? Am J Epidemiol. 110(1979): 77-87.
- Nichols AB, Ravenscroft C, Lamphiear DE, et al. Independence of serum lipid levels and dietary habits. JAMA 236(1976): 1948-1953.
- Noakes TD. 1996 JB Wolffe Memorial Lecture. Challenging beliefs: ex Africa semper aliquid novi. Med Sci Sports Exerc. 29(1977): 571-590.
- Ramsden CE, Zamora D, Leelarthaepin B, et al. Use of dietary linoleic acid for secondary prevention of coronary heart disease and death. Evaluation of recovered data from the Sydney Diet Heart Study and updated meta-analysis. BMJ 346(Feb. 4, 2013): e8707.
- Taubes G. Good Calories, Bad Calories: Fats, Carbs, and the Controversial Science of Diet and Health. New York, NY: Anchor Books, 2008.
- Castelli WP. Concerning the possibility of a nut. Arch Intern Med. 152(1992): 1371-1372.
- Castelli WP, Davidson M, Connor WE, et al. Current Concepts in Cardiology. Morris Plains, NJ: Warner-Lambert, 1987. Cited by Enig MG. Diet, serum cholesterol, and coronary heart disease. In: Coronary Heart Disease. The Dietary Sense and Nonsense. An Evaluation by Scientists. Mann GV Ed. London, England: Janus Publishing Company, 1993. pp. 37-60.
- Keys A, Mickelsen O, Miller EVO. The relation in man between cholesterol levels in the diet and in the blood. Science 113(1950): 79-81.
- Keys A, Anderson JT, Mickelsen O, et al. Diet and serum cholesterol in man: Lack of effect of dietary cholesterol. J Nutr. 59(1956): 39-56.
- Keys A. Letter: Normal plasma cholesterol in a man who eats 25 eggs a day. N Engl J Med. 325(1991): 584.
- Boffey, PM. Cholesterol: debate flares over wisdom in widespread reductions. New York Times, 14 July 1987.
- Kannel WB, Dawber T, Kagan A, et al. Factors of risk in the development of coronary heart disease – Six-year follow-up experience. The Framingham Study. Ann Intern Med. 55(1961): 33-50.
- Dawber TR, Moore FE, Mann GV. II. Coronary heart disease in the Framingham study. Am J Public Health 47(1957): 4-24. Republished Int J Epidemiol. 44(2015): 1767-1780.
- Kannel WB. Metabolic risk factors for coronary heart disease in women: Perspective from the Framingham Study. Am Heart J. 154(1987): 413-419.
- Kannel WB. Lipids, diabetes, and coronary heart disease: Insights from the Framingham Study. Am Heart J. 110(1985): 1100-1107.
- Kannel WB, Castelli WP, Gordon T. Serum cholesterol, lipoproteins, and the risk of coronary heart disease. The Framingham Study. Ann Intern Med. 74(1971): 1-12.
- Kannel WB, McGee DL. Diabetes and cardiovascular risk factors: The Framingham Study. Circulation 59(1979): 8-13.
- Smith RL, Pinckney ER. The Cholesterol Conspiracy. St. Louis, Missouri: Warren H. Green Inc., 1991.
- Kannel WB, Dawber TR, Friedman GD, et al. Risk factors in coronary heart disease. An evaluation of several serum lipids as predictors of coronary heart disease. The Framingham Study. Ann Intern Med. 61(1964): 890-899.
- Castelli WP, Anderson K. A population at risk. Prevalence of high cholesterol levels in hypertensive patients in the Framingham Study. Am J Med. 80(1986, suppl 2A): 23-32.
- Castelli WP. Lipids, risk factors and ischemic heart disease. Atherosclerosis 124(1996): S1-S9.
- Grundy SM. Cholesterol and coronary heart disease. A new era. JAMA 256(1986): 2849-2858.
- Livshits G, Weisbort J, Meshulam N, et al. Multivariate analysis of the twenty-year follow-up of the Donolo-Tel Aviv Prospective Coronary artery disease study and the usefulness of high density lipoprotein cholesterol percentage. Am J Cardiol. 63(1989): 676-681.
- Stamler J, Wentworth D, Neaton JD. Is relationship between serum cholesterol and risk of premature death from coronary heart disease continuous and graded? Findings in 356 222 primary screenees of the Multiple Risk Factor Intervention Trial (MRFIT). JAMA 256(1986): 2823-2828.
- World Health Organization. Prevention of coronary heart disease. Report of a WHO Expert Committee, Geneva, 30 Nov.-8 Dec. 1981. Technical Report Series. World Health Organization 678(1982): 53.
- Whyte HM. Potential effect on coronary-heart-disease morbidity of lowering the blood-cholesterol. Lancet 305(1975): 906-910.
- Taylor WC, Pass TM, Shepard DS, et al. Cholesterol reduction and life expectancy: A model incorporating multiple risk factors. Ann Intern Med. 106(1987): 605-614.
- Browner WS, Westenhouse J, Tice JA. What if Americans ate less fat? A quantitative estimate on the effect on mortality. JAMA 265(1991): 3285-3291.
- Grover SA, Gray-Donald K, Joseph L, et al. Life expectancy following dietary modification or smoking cessation. Estimating the benefits of a prudent lifestyle. Arch Intern Med. 154(1994): 1697-1704.
- Gordon T, Castelli WP, Hjortland MC, et al. High density lipoprotein as a protective factor against coronary heart disease. The Framingham Study. Am J Med. 62(1977): 707-714.
- Kannel WB, Neaton JD, Wentworth D, et al. Overall and coronary heart disease mortality rates in relation to major risk factors in 325,348 men screened for the MRFIT. Am Heart J. 112(1986): 825-836.
- Cutler JA, Neaton JD, Kuller L, et al. Coronary heart disease and all-cause mortality in the Multiple Risk Factor Intervention Trial: subgroup findings and comparison with other trials. Prev Med. 14(1985): 293-311.
- Iso H, Jacobs DR, Wentworth D, et al. Serum cholesterol levels and six-year mortality from stroke in 350,977 men screened for the Multiple Risk Factor Intervention Trial. N Engl J Med. 320(1989): 904-910.
- U.S. Senate Select Committee on Nutrition and Human Needs. Dietary Goals for the United States, 2nd ed. Washington, DC, U.S. Government Printing Office, 1977. (See also here).
- Nair P. Brown and Goldstein: The cholesterol chronicles. PNAS 110(2013): 14829-14832.
- Brown MS, Goldstein JL. Familial hypercholesterolemia: Defective binding of lipoproteins to cultured fibroblasts associated with impaired regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity. PNAS 71(1974); 788-792.
- Goldstein JL, Brown MS. Familial hypercholesterolemia. A genetic regulatory defect in cholesterol metabolism. Am J Med. 58(1975): 147-150.
- Brown MS, Goldstein JL. A tribute to Akira Endo, discoverer of a “Penicillin” for cholesterol. Ather. Suppl. 5(2004): 13-16.
- Brown MS, Goldstein JL. Lowering plasma cholesterol by raising LDL receptors. N Engl J Med. 305(1981): 515-517.
- Bilheimer DW, Grundy SM, Brown MS, et al. Mevinolin stimulates receptor-medicated clearance of low density lipoprotein from plasma in familial hypercholesterolemia heterozygotes. Trans Assoc Am Physcns. 96(1983): 1-9.
- Lipid Research Clinics. Lipid Research Clinics Coronary Primary Prevention Trial results.I. Reduction in incidence of coronary heart disease. JAMA 251(1984): 351-364.
- Lipid Research Clinics. Lipid Research Clinics Coronary Primary Prevention Trial results. 11. The relationship of reduction in incidence of coronary heart disease to cholesterol lowering. JAMA 251(1984): 365-374.
- Ware WR, The mainstream hypothesis that LDL cholesterol drives atherosclerosis may have been falsified by non-invasive imaging of coronary artery plaque burden and progression. Med Hypotheses 73(2009): 596–600.
- Robins SJ, Lyass A, Zachariah JP, et al. Insulin resistance and the relationship of a dyslipidemia to coronary heart disease. The Framingham Heart Study. Arterioscler Throm Vasc Biol. 31(2011): 1208-1214.
- Cutter JA, Neaton JD, Hulley SB, et al. Coronary heart disease and all-causes mortality in the Multiple Risk Factor Intervention Trial: subgroup findings and comparisons with other trials. Prev Med. 14(1985): 293-311.
- Stamler J, Neaton JD, Cohen JD, et al. Multiple Risk Factor Intervention Trial revisited: A new perspective based on nonfatal and fatal composite endpoints, coronary and cardiovascular, during the trial. J Am Heart Assoc. 1(2012): e003640.
- Strandberg TE, Salomaa VV, Naukkarinen VA, et al. Long-term mortality after 5-year multifactorial primary prevention of cardiovascular disease in middle-aged men. JAMA 266(1991): 1225-1229.
- Noakes TD. It’s the insulin resistance, stupid: Part 5. CrossFit.com. 22 Oct. 2019. Available here.
- Kealey T. Why does the Federal Government issue damaging dietary guidelines? Lessons from Thomas Jefferson to today. Cato Institute Policy Analysis No 846. Available here.
- Lackland DT, Roccella EJ, Deutsch AF, et al. Factors influencing the decline in stroke mortality. A statement from the American Heart Association/American Stroke Association. Stroke 45(2014): 315-353.
- Howard BV, Van Horn L, Hsia J, et al. Low-fat dietary pattern and risk of cardiovascular disease. The Women’s Health Initiative Randomized Controlled Dietary Modification Trial. JAMA 295(2006): 655-66.
- Noakes TD. The Women’s Health Initiative Randomized Controlled Dietary Modification Trial: An inconvenient finding and the diet-heart hypothesis. S Afr Med J. 103(2013): 824-825.
- Noakes TD, Sboros M. Real Food on Trial. How the Diet Dictators Tried to Destroy a Top Scientist. U.K.: Columbus Publishing Ltd, 2019.
- Mann GV. The Way It Seems. Los Angeles, CA: Vantage Press, 1988.
- Kannel WB, Dawber TR, Cohen ME, et al. Vascular disease of the brain – epidemiologic aspects: The Framingham Study Am J Publ Health 55(1965): 1355-1366.
- Kannel WB, Wolf PA, Verter J, et al. Epidemiologic assessment of the role of blood pressure in stroke. The Framingham Study. JAMA 214(1970): 301-310.
- Kannel WB, Castelli WP, McNamara PM, et al. Role of blood pressure in the development of congestive heart failure. The Framingham Study. N Engl J Med. 287(1972): 781-787.
- McKee PA, Castelli WP, McNamara PM et al. The natural history of congestive heart failure: The Framingham Study. N Engl J Med. 285(1971): 1441-1446.
- Pett KD, Kahn J, Willett WC, Katz DL. Ancel Keys and the Seven Countries Study: An evidence-based response to revisionist histories. White Paper commissioned by The True Health Initiative. 1 Aug. 2017. Available here.
- Willett W. Challenges for public health nutrition in the 1990s. Am J Publ Health. 80(1990): 1295-1298.
- Willett W. Nutritional Epidemiology. New York: Oxford University Press, 1990.
- Shekelle RB, Shryock AM, PaulO. Diet, serum cholesterol and death from coronary heart disease. The Western Electric Study. N Engl J Med. 304(1981): 65-70.
- Keys A. Seven Countries: A Multivariate Analysis of Death and Coronary Heart Disease. Cambridge, MA: Harvard University Press, 1980.
- Grundy SM. Trans mono unsaturated fatty acids and serum cholesterol levels. N Engl J Med. 323(1990): 480-481.
- Mann GV. The clinical trials. In: Coronary Heart Disease. The Dietary Sense and Nonsense. An Evaluation by Scientists. Mann GV Ed. London, England: Janus Publishing Company, 1993. pp.61-75.