1959: Albrink and Man Claim Hypertriglyceridemia Explains the Development of CHD in Persons With T2DM
My previous articles on insulin resistance began (2) with the story of how Drs. Margaret Albrink and Evelyn Man identified elevated blood triglyceride concentrations as a more probable risk factor for CHD than the blood cholesterol concentration. This insight then inspired research into the mechanism of hypertriglyceridemia and the identification of carbohydrate-sensitive hypertriglyceridemia (2).
Albrink and Man were fairly certain the dietary cause of CHD was more likely to be carbohydrates than saturated fats (3-6), Albrink held this position through the mid-1960s when she published her final editorial (6).
There, she wrote:
The association between impaired carbohydrate metabolism and atherosclerosis reported by Ostrander and associates (7) and between dietary carbohydrate and hyper(tri)glyceridemia reported by Kuo and Bassett (8) are consistent with hypotheses that the common modern diseases of diabetes, atherosclerosis, and obesity and associated hyperglyceridemia may be the present day manifestations of the effect of affluence on a once useful genetic trait, the ability to conserve carbohydrate. (7, p. 1332)
However, as I describe subsequently (9, 10), this was the last occasion that anyone would be allowed to promote the idea that carbohydrates could be, in any way, implicated as a cause of CHD. Nor, going forward, could anyone suggest that a genetic trait — perhaps insulin resistance or “the ability to conserve carbohydrate” — might be involved.
Industry, in the form of the U.S. Sugar Research Foundation, ensured Albrink and Man’s evidence-based theory would be quickly written out of history.
1960: The NHI Establishes Executive Committee on Diet and Heart Disease, Proposes a National Diet-Heart Study
Even as the American Heart Association (AHA) was describing Keys’ hypotheses as speculative and unproven, in 1960 the National Heart Institute (NHI) formed an Executive Committee on Diet and Heart Disease under the chairmanship of Dr. Irvine Page, who had been the lead author of its 1957 statement on atherosclerosis and fat content in the diet (11).
The committee — the composition of which I could not find but which may have included Ancel Keys, Benjamin Baker, Jeremiah Stamler, Fredrick Stare, Laurance Kinsell, and/or Ivan Frantz — decided that a large clinical trial was needed to test the hypothesis that “among middle-aged American men, alteration of the amount of and type of fat and amount of cholesterol in the diet would decrease the incident of future clinical coronary heart disease” (12, p. 105). It further argued that the most appropriate intervention would be a dietary modification independent of any other changes in the lifestyles of the participants. The conclusion was that a trial following 100,000 men for four to five years would be needed to detect a 20% reduction in CHD events. Since this would be a massive undertaking costing perhaps a billion dollars, the committee recommended rather a smaller-scale, feasibility study. Thus, the cooperative two-year Feasibility Trial of the National Diet-Heart Study (FTNDHS) was launched (13, p. 131).
Importantly, the funding of this exclusive group of scientists, all of whom were infected with the virus of confirmation bias, produced obvious conflicts of interests that were not then recognized: The very people whose careers would benefit the most from the diversion of taxpayer money to the study of CHD were exactly the people entrusted to decide whether such studies should be attempted, where and by whom those studies should be conducted, how much money should be invested in each of them, and most importantly, who would analyze the data and write up the scientific manuscripts. The FTNDHS had become a classic example of the foxes guarding the hen house.
The stated goal of the study was to determine whether it was feasible to study free-living humans, two groups of which would replace most of the saturated fat and cholesterol in their diets with polyunsaturated fatty acids (PUFAs). The principal investigators were to be Drs. Baker, Keys, Frantz, Kinsell, Stamler, and Stare. Interestingly, the only person on the team who did not have medical training was Keys himself. This grouping of laboratories came to be known as the diet-heart centers. In time additional laboratories joined to form the 12 laboratories involved in the Lipid Research Clinics Coronary Primary Prevention Trial, discussed subsequently.
The inquisitive reader will soon realize this group inevitably would become Keys’ greatest supporters and the primary beneficiaries of funding from the AHA-NIH alliance. Keys must have known this tight group of friends was clever enough to understand their mandate. They would never, as the saying goes, be stupid enough to bite the hands that fed their voracious appetites. Rather, any support they might offer to bolster Keys’ hypotheses would only enhance their own careers by increasing the probability of increased and sustained funding from the developing alliance between the AHA, NHI, and U.S. government.
So predictably, they chose not to give equal time to studies of other possible dietary interventions — for example, the use of carbohydrate restriction to treat carbohydrate-sensitive hypertriglyceridemia (CSHT).
Nor did they ever pretend to test the null hypothesis, which is sacred to the scientific process if one wishes to avoid confirmation bias: One designs the trial on the assumption the intervention one wishes to test will not produce a positive outcome. Instead, this band of scientific brothers interpreted all their findings according to their preexisting confirmation bias. Trials that failed to spawn favorable outcomes in support of the diet-heart hypothesis, as almost all would do over the next 30 years, were explained away as if each was the sole unfortunate outlier and safely could be ignored.
In their unified certainty that saturated fat causes CHD and CHD can be prevented by a low-fat diet, they simply ignored the concept of the null hypothesis.
In announcing the National Diet-Heart Study to the world, Baker, Keys, Kinsell, Page, Stamler, and Stare expressed absolute certainty about what they were destined to discover: “Much evidence implicates diet as one of the key etiological factors (causing atherosclerosis). … Within the last 10 years several investigators have undertaken studies to examine the influence of dietary modification on the occurrence of clinical coronary heart disease” (12, p. 105).
These statements are hubristic and neither is factually correct. All the available evidence at the time was purely circumstantial, and there were no studies of the influence of dietary interventions specifically on CHD endpoints. These false statements were merely used to justify the need for “a national cooperative effort … to accomplish this difficult research mission” (p. 105), an “effort” that would dramatically enhance the research careers of these ambitious researchers, even if it failed to advance human understanding of what causes CHD.
Left unstated was the nature of the mission: Was it to determine whether diet plays a significant role in the development of atherosclerosis? Or was it to provide the missing evidence necessary to “prove” what Keys and his acolytes had already decided was a biblical truth?
The design of the trial required that 1,500 healthy adult males between ages 45 and 54 be recruited for a 13-month study, during which different groups would receive foods “known to lower serum cholesterol levels,” which was “suggested as a possible way of preventing coronary heart disease” (12, p. 106). Participants were instructed to purchase fat-containing foods from special food centers established by the study directors. Food orders were to be submitted weekly and were then delivered to the participants’ homes.
Interestingly, the food industry was listed as a crucial collaborator in the study, as were heart associations, medical societies, and many governmental agencies (p. 106).
When that exploratory study (12, 13) was duly completed, it identified a number of challenges. First it was difficult to recruit subjects, as only between 5-10% of persons invited to participate ultimately consented. Second, a large number of persons recruited for the study dropped out for a variety of reasons. Third, preparation of the different diets proved to be challenging. Worse, 44% of subjects adhered poorly or only fairly well to the diet (14, 15).
Despite this, blood cholesterol concentrations fell in those eating the diets lower in cholesterol and saturated fats, but the reduction was not large, ranging from 11 to 17%. However, the better the dietary compliance, the greater the reduction in blood cholesterol concentrations (15). Subjects also lost some weight (4-5%) for the first 12 weeks of the trial, after which there was some weight regain (13).
Predictably, despite these marginal outcomes, the foxes (authors) ultimately argued a larger study of approximately 60,000 men aged 40-59 from 20 large cities across the U.S. was required (13). By now they realized the study could not be double-blinded so that neither the foxes nor the hens knew which diet specific participants were eating. It was difficult to motivate subjects to adhere to the alternative diet, the dietary intervention: “Motivation of participants was difficult to effect under the double-blind requirement. There were no tangible goals for nutritionists or participants because diet composition and serum cholesterol levels were not known to them” (13, p. 142).
But without a double-blind intervention, the trial was biased toward a more favorable outcome in the intervention group, because both the foxes and the hens know the goal of the trial is to produce better outcomes in the intervention group. That knowledge inevitably would play a large part in determining the final outcome of the trial.
Even though this exploratory study added nothing to the state of knowledge in 1968, Helen Brown and Irvine Page did not waste any time in promoting their position that this research program was very important:
While no one thinks diet alone is the keystone to the control of coronary disease, enough evidence has already been published by investigators … to suggest that it may be helpful. The problem is too important to neglect … . The time has surely come when the search for new ideas and the validation of old ones are the paramount issues in the control of heart attacks. From such studies will come the “practical” procedures applicable to any control program for coronary disease. (14, pp. 314-315, my emphasis)
The same authors elsewhere wrote:
Again physicians and public health administrators are challenged by the extremely high incidence and mortality of coronary heart disease. Preventive measures are imperative. Recent reports on the close association between dietary change (and serum cholesterol reduction) and the occurrence of coronary heart disease are encouraging. But these reports concern relatively small numbers of men and do not solve the medical and public health problem. Consequently, the large study designed to give a definitive answer to the problem should be conducted soon. (13, p. 142)
They also wrote:
This in no way implies that diet is the only important factor in the genesis of atherosclerosis, but since this study had demonstrated the feasibility of significantly modifying the diet of a free living population, it is now imperative to quantitate the magnitude of the potential saving of life by such dietary alterations. Many lines of evidence suggest that it will be considerable. (16, p. 474, my emphasis).
What is remarkable about all these statements is that each establishes the overwhelming intellectual bias the key researchers brought to their scientific endeavors. Their overpowering desire was to prove that a particular diet that lowers the blood cholesterol concentration is the single most important factor in determining future risk of heart attack.
Science is expected to be neutral, to be conducted by those who have no material or intellectual interest in what will be the outcome. Science is meant to expose the truth, whatever that truth might be.
It is absolutely clear that all the AHA/NHI-funded research of Keys’ hypotheses was conducted solely by those who were inexorably biased toward finding support for them. That bias was strengthened by industry and other organizations that so willingly participated in this initial trial. Their influence was substantial.
Nina Teicholz (17) claims the National Diet-Heart Study “could reasonably be viewed in part as an industry-driven effort to broaden the market for its commodity oil.” She continues, “Companies contributing to the study included nearly every major food corporation in the country including the vegetable oil giant Anderson, Clayton & Company, Carnation, The Corn Products Company, Frito-Lay, General Mills, H.J. Heinz, the Pacific Vegetable Oil Corporation, Pillsbury, and Quaker Oats, among others” (17, p. 91).
These companies skillfully directed popular opinion away from the question everyone should have asked: “How could it be that a healthy diet would depend upon those just-invented foods, such as milk ‘filled’ with soybean oil?” (17, p. 81).
This was the question Procter and Gamble so skillfully had avoided when in 1910 they marketed Crisco as the healthier alternative to the animal-derived fats we and our hominid ancestors had been eating for millions of years.
It is a question about which the general public continues to be misinformed as we move increasingly toward the era in which these invented foods dominate all that we eat.
1961: AHA Begins Advising That Dietary Saturated Fat Be Replaced With Polyunsaturated Fatty Acids (PUFA)
In a remarkable turnabout that was not based on the recent appearance of any new knowledge, in 1961 the AHA suddenly decided, oh wait a moment, we were wrong. Keys, it now determined, had been right all along.
In a three-page summary published in February 1961 (18), the AHA essentially reversed its far more detailed and rigorously argued 1957 declaration (11). Whereas in 1957 the AHA had argued there was insufficient evidence to support Keys’ hypotheses, now just four years later, there was quite suddenly enough evidence to support a definitive change in U.S. eating habits, specifically to prevent CHD.
The report developed its argument in fairy tale fashion as if it had been written as an advertorial for the AHA by a Madison Avenue public relations company. Clearly it was not about science. Thus, the AHA report stated:
Many years ago (in a fairyland inhabited by pixies and goblins) a scientist fed cholesterol and other types of fat to rabbits. The blood cholesterol increased and the rabbits developed atherosclerosis. … Global studies have shown that dietary habits of human populations differ. Evidence gathered from many countries suggests a relationship between the amount and type of fat consumed, the amount of cholesterol in the blood, and the reported incidence of coronary artery disease. (18, p. 389, my irreverent addition and emphasis)
The report claimed:
These and other research studies have given clues to the prevention of atherosclerosis by dietary means. A reduction in the blood cholesterol by dietary means, which also emphasizes weight control, may lessen the development or extension of atherosclerosis and hence the risk of heart attacks or strokes. (p. 389, my emphasis)
The AHA’s advisory also included a section describing the type of diet that would produce these miraculous outcomes, even though no study had then — or even now — shown any effect of a dietary intervention on CHD all-cause mortality:
The blood cholesterol concentration may also be reduced by controlling the amount and type of fat in the diet without altering caloric intake. Not all fats in the diet have the same effect on the amount of cholesterol in the blood. In the usual diet eaten in the United States, a large part of the fat is of the saturated fat type. Too much of this type of fat tends to increase the cholesterol in the blood. Considerable amounts of saturated fat are present in whole milk, cream, butter, cheese, and meat. Coconut oil and the fat in chocolate also have a high content of fats of the saturated type. Most shortenings and margarines have less than half as much saturated fat, and the common vegetable oils have still less. When the intake of saturated fats is reduced, blood cholesterol levels usually decrease.
In contrast to the above food fats, many natural vegetable oils such as corn, cotton and soy as well as the fat of fish, are relatively low in saturated fats and high in fats of the poly-unsaturated type. When these fats are substituted for a substantial part of the saturated fats without increasing calories, blood cholesterol decreases. Finally, some food fats such as olive oil are intermediate in saturation and have no strong effect one way or the other on the blood cholesterol.
These measures make it possible to attempt a considerable alteration in the cholesterol level in the blood with the use of acceptable diets. …
In conclusion — The reduction or control of fat consumption under medical supervision, with reasonable substitution of poly-unsaturated fats for saturated fats, is recommended as a possible means of preventing atherosclerosis and decreasing the risk of heart attacks and strokes. This information is based on the best scientific information available at the present time. (p. 390, my emphasis)
Of course this is nonsense, since the “best scientific information available” in 1961 was no different from the same “best evidence” available in 1957 when a much more thorough review written by Page, Stare, Corcoran, Pollock, and Wilkinson and not by the paid writers of commercial advertorials had concluded there was no such credible evidence (11).
Indeed, the report included the disclaimer: “It must be emphasized that there is as yet no final proof that heart disease or strokes will be prevented by such measures” (18, p. 389). (As I will argue, that statement remains valid today, 59 years later)
But things had changed at the AHA between 1957 and 1961. The authors of the 1961 report were now Page, Stare, Allen, and crucially, Keys and Stamler. So Keys and Stamler — whose commitment to the cause would earn him an appointment as director of the hugely expensive MRFIT study (19) — had been added to the AHA committee. It was perhaps natural that Keys and Stamler would be keen to steer the committee to find in favor of Keys’ unproven and still untested hypotheses. Each had so much to gain from this novel consensus.
As Teicholz explains, at the time Stamler was also in the process of cozying up to the vegetable oil industry in order to secure funding for his research:
Campbell Moses, AHA medical director in the late 1960s, even posed with a bottle of Crisco Oil in an AHA educational film. And remarkably, when Jerry Stamler reissued his 1963 book, Your Heart Has Nine Lives, it was published as a “professional” red leather edition by the Corn Products Company and distributed free of charge to thousands of doctors. Inside, Stamler thanks both that company and the Wesson Fund For Medical Research for “significant” research support. “Scientists in public health must make alliances with industry” he told me unashamedly when I asked him about the connection. “It’s tough.” (17, pp. 85-86)
In effect, Stamler had sold out to the seed (“vegetable”) oil industry and was no longer an independent, disinterested scientist searching for an ultimate dietary truth. Instead, his attachment to “healthy polyunsaturated fats” was irrevocable, as would become the case for vegan/vegetarian activists Walter Willett and Frank Hu at the Harvard T. H. Chan School of Public Health (20) and Dariush Mozafarrian (21) at the Tufts Friedman School of Nutrition Science and Policy 30 years later. The research of all these leading nutritional scientists would, like that of Stamler, become heavily dependent on funding from the “vegetable” oil industry (22) and various front organizations like the International Life Sciences Institute (23-25).
Willett’s role in the development and promotion of his still unproven Mediterranean diet is fully described by Teicholz (17, pp. 174-224), as are the roles of industry and the Greek and Italian authorities. Her conclusions are not sanguine:
In fact, one of the more disturbing aspects of the Mediterranean diet pyramid is that it has intensified America’s phobia about animal fats, accelerating our flight from these ancient foods to using vegetable oils instead. And this result may have harmed health in ways that appear serious but have not yet been well researched — because experts have for so long been focused exclusively on the supposed dangers of eating meat and dairy instead. (17, p. 224)
I’ve made the point before (26) that it took 30 years for the damaging effects of trans fats produced in the original hydrogenation process used to convert “vegetable” oils to solid margarines and shortening (for use in baking) to finally be acknowledged. Yet there are no data to show the processes currently used to produce vegetable oils with fewer or no trans fats are not just as harmful or perhaps even more so. All we have are the statements of industry-directed scientists urging us to avoid saturated fats and to ingest polyunsaturated “vegetable” oils. It seems we have not learned the lesson from the trans fats debacle (17), which is a predictable outcome if industry controls the messaging that leading scientists are providing.
In his relentless and uncompromising support of “vegetable” oils, Mozafarrian, who by some reports wishes to be known as the next Great Man of Nutrition, has two major problems. First, he is heavily conflicted, having been the recipient of funding from a wide range of commercial companies, which until recently included Unilever (22, 27). Second, he has published an article (28) showing that unlike saturated fats, both carbohydrates and polyunsaturated fats are associated with the progression of coronary atherosclerosis in elderly postmenopausal women (Figure 1).
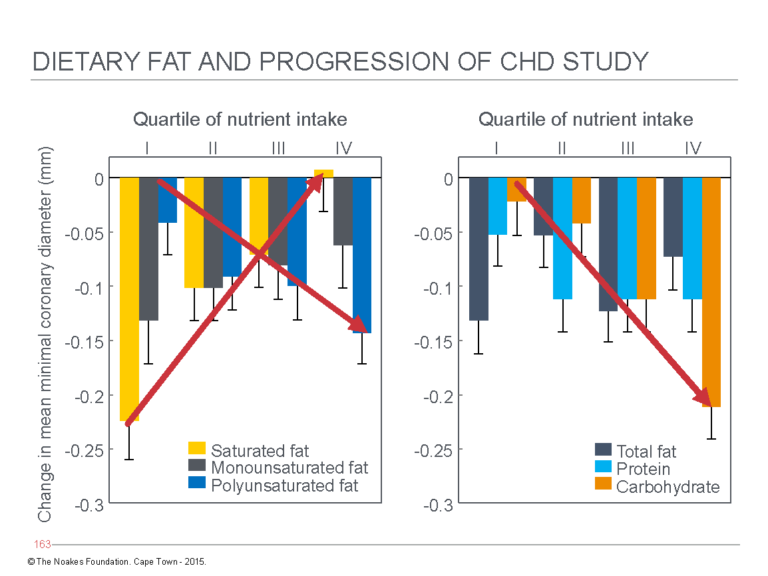
Figure 1: Changes in coronary artery narrowing — a measure of progression of coronary atherosclerosis — in postmenopausal women eating their usual diets. The arrows indicate change in coronary artery narrowing with increasing consumption of saturated fats or polyunsaturated fats (“vegetable” oils) (left panel) or of carbohydrates (right panel). The left panel shows women who ate the least saturated fat (yellow column on extreme left) showed the greatest progression in coronary atherosclerosis, whereas those who ate the most had the least progression. Disease progression was also accelerated in those eating more polyunsaturated fats (blue columns). The right panel shows accelerated disease progression in women reporting higher carbohydrate intakes (orange columns). Mozaffarian, the senior author of this study, continues to promote the value of diets high in polyunsaturated fats and carbohydrates as the healthiest option. This is known as cognitive dissonance. Reproduced from Figure 1 in reference 28.
The study the coronary artery diameters of 235 women were measured before and after a three-year period, during which they participated in a drug trial to determine whether hormone replacement therapy would influence the progression of coronary atherosclerosis in postmenopausal women. Women completed a dietary questionnaire at the beginning of the trial, which recorded their intakes of carbohydrate; protein; and saturated, monounsaturated, and polyunsaturated fats. Subjects were classified into quartiles based on their intakes of these macronutrients.
Coronary angiography was performed before and after the three-year interval to determine progression of coronary atherosclerosis. The results depicted in Figure 1 clearly show minimal progression in those eating the most saturated fat. In contrast, high intakes of either carbohydrates or polyunsaturated fats were associated with the most rapid progression. The result of this hugely expensive and rigorously conducted trial could not have been clearer (at least to those unaffected by serious conflicts of interest).
The authors — and this naturally included Mozaffarian as the senior author — concluded: “In postmenopausal women with relatively low total fat intake, a greater saturated fat intake is associated with less progression of coronary atherosclerosis, whereas carbohydrate intake is associated with greater progression” (28, p. 1175).
Not so fast, Dr. Dariush. Did you somehow fail to notice (Figure 1; arrow in left panel) that progression was also increased in women who ate the most polyunsaturated fat? Oh, I almost forgot. Your sponsors would not like you to write anything so negative about their product.
But to return to the 1961 AHA document, one outcome is certain: The AHA in 1961 would not risk funding any research that might jeopardize its dependence on the largesse of the food industry. Its mission had become to stay in business, not to make itself irrelevant (by actually preventing heart disease).
So with Keys and Stamler’s support and the influence of the food industry in the foreground, it is not surprising that the 1961 AHA statement included this advice:
The reduction or control of fat consumption under medical supervision, with reasonable substitution of poly-unsaturated fats for saturated fats, is recommended as a possible means of decreasing the risk of heart attack or stroke. The recommendation is based on the best scientific information available at the present time. (18, p. 390)
Of course, this is disingenuous, because there was no “best scientific information” at the time, just best speculation (as is the case today). Instead, the 1961 AHA report was nothing more than a paid advertisement created by a financial transaction between the AHA and, at the time, its premier sponsors in the food industry.
In plain English, it was simply a bribe — the first of many.
While the report would produce a highly beneficial return on investment for the food and, ultimately, the medical industries (as a result of the rise in sick people globally needing care), its abiding legacy is the obesity/diabetes pandemic that it unleashed.
1962-2016: NHI grants Keys, Stamler, and Frantz Funding for the Minnesota Coronary Experiment (MCE)
As part of its funding allocated for the National Diet-Heart Study, a special additional allocation was made to Keys, Stamler, and Frantz for the initiation of the Minnesota Coronary Experiment (MCE) (29), which began in November 1968.
In a previous column (30), I reviewed the MCE in some detail and introduced the critical finding from the recovered MCE (RMCE) study (31), which will be discussed in more detail in a future column (32).
The historical relevance of the MCE, and more especially the RMCE, is that it established that the replacement of saturated fat in the diet causes measurable harm. As a result, if the MCE had been properly analyzed and published in full, as it should have been in 1976, the findings would have made it unethical for any future study to evaluate the effects of removing saturated fat from the diet. It would be unethical because the removal of saturated fat from the diet in the intervention group in the MCE clearly caused significant harm to many, and on the ethical principle that the first rule in medicine is to “do no harm,” any study of an intervention already shown to cause harm is unethical.
In particular, the Women’s Health Initiative Randomized Controlled Dietary Modification Trial (WHIRCDMT) (33) could not ever have been justified, because the RMCE proved the only possible outcome of the WHIRCDMT would be harm to the group randomized to the low-fat dietary intervention. This was exactly what happened, as postmenopausal women with established heart disease or T2DM at the start of the trial suffered harm when they reduced their intakes of dietary saturated fats (34). I will discuss this in greater detail subsequently (35).
1965: Hegsted and Stare Health-Wash Sugar and Demonize Saturated Fats
July 1, 1965, was perhaps the single most important day in the history of the evolution of Keys’ cholesterol scam.
On that day, John Hickson, Vice President and Director of Research at the Sugar Research Foundation (SRF) — now known as the Sugar Association — paid a special visit to the Nutrition Department at the Harvard University School of Public Health (36-38). Stare, the Head of the Department, was highly connected to industry and influential organizations, including the AHA, the NHI, and the National Academy of Sciences (39). Stare was clearly partial to using his scientific gravitas to drive industry’s commercial interests (40, 41). Already a member of the SRF Scientific Advisory Board, Stare was clearly a prize target for pushing the sugar industry’s global agenda, so the goal of Hickson’s visit was likely to encourage Stare and his Harvard colleagues to help the SRF in its campaign to health-wash sugar of any possible blame in causing CHD. This would mean demonizing saturated fat as the key driver.
In December 1964, Hickson had reported to the SRF that evidence supporting a potential role of sugar in the cause of ill health was escalating alarmingly. He reported: “From a number of laboratories of greater or lesser repute, there are flowing reports that sugar is a less desirable source of calories than other carbohydrates, e.g., [John] Yudkin” (36, p. 1682). He proposed the SRF “could embark on a major program” to counter Yudkin and other “negative attitudes to sugar” (p. 1682). Central to this campaign would be the use of credible scientists (like Stare and his trusted deputy Mark Hegsted), working at the most trusted academic institutions to write articles, appropriately placed in the most prestigious medical and scientific journals, that would “refute our detractors” (p. 1682).
As Hickson climbed the steps to the Harvard School of Public Health on that warm New England summer’s day, he carried in his briefcase copies of four articles recently published in the June 1965 issue of the Annals of Internal Medicine. The four articles were especially alarming to the SRF, since they all suggested sucrose (sugar) might have a special role in the causation of CHD.
The first article (42) was a product of the Tecumseh Community Health Study, which found blood cholesterol concentrations were unrelated to dietary factors in the Tecumseh population (43-45). Interestingly, despite the message he would convey in this article, senior author Frederick Epstein would ultimately become one of Keys’ closest allies and the staunchest possible supporter of the findings of the Seven Countries Study (46). By then his bias had become blatant, but in 1965 he was clearly still able to see the evidence for what it was. Why he subsequently underwent a dramatic reversal in his opinion has yet to be uncovered. I can offer some guesses.
The data from the Tecumseh Study found an elevated blood glucose concentration, even in the absence of T2DM, was at least as good a predictor of future CHD risk as was the blood cholesterol concentration (42).
The second study (47), also from the Tecumseh Community Health Study, was one of the first to evaluate the total extent of arterial disease throughout the body in persons with T2DM. It found the presence of T2DM predicted more severe disease in all the arteries throughout the body, not just in the coronary arteries: “The detection of hyperglycemia with significant frequency among persons with each of five different manifestations of vascular disease is a unique finding among epidemiologic studies” (p. 1195). The study authors also noted, “A high prevalence of hyperglycemia has not been reported among persons with vascular disease in any previous epidemiological study” (p. 1196).
The senior author of the third study, Dr. Peter Kuo (48), had been a key early promoter of the concept of carbohydrate-induced hyper(tri)glyceridemia as the most common finding in patients with CHD (49; Figures 6 and 7 in reference 50). In their paper, Kuo and Drew Bassett compared the effects of dietary carbohydrate as either sugar or starch on blood lipid levels in individuals (Figure 2) and in groups.
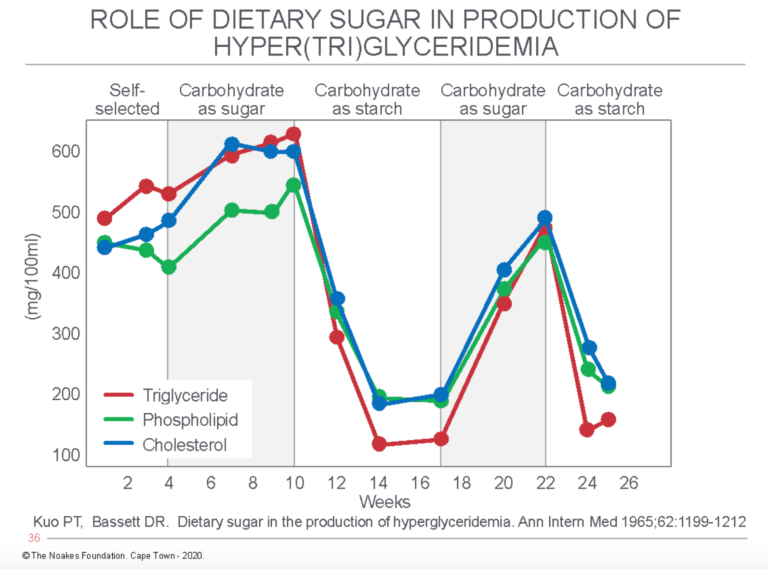
Figure 2: The effects of dietary carbohydrate as either sugar or starch on blood concentrations of triglycerides, phospholipids, and cholesterol. Note that sugar increases, whereas starch reduces the blood concentrations of all three lipids. Reproduced from reference 48.
The authors concluded: “The data indicate that sugar is a potent lipemic agent as compared with starches, and a high dietary sugar intake may well be the most important factor in the production of the clinically encountered type of hyperglyceridemia” (48, p. 1199).
The final paper (51) in the series was an editorial by Albrink, who had pioneered the concept that hypertriglyceridemia is more prevalent than hypercholesterolemia in persons with CHD (50, 52).
In the editorial, Albrink argued carbohydrate was a relatively recent addition to the human diet:
Not until the advent of agriculture 8,000 to 10,000 years ago did carbohydrate in the diet become copiously available. By the time agriculture was introduced man had already evolved to his present physical shape, and very likely, therefore to his present biochemical, hormonal, and enzymatic form. (51, p. 1330)
But humans had evolved as carnivorous hunters who relied on stored body fats for energy. The human brain, however, still required some glucose. Thus Albrink wrote, “Primitive man could cope with his life only if he were able to store reserve calories as fat to be called upon as fuel for the rest of the body in times of muscular effort or starvation. The mechanisms whereby such conservation of carbohydrate and ready utilization of fat are brought about are now beginning to be understood” (p. 1331). Importantly, the habitual diet of early humans promoted carbohydrate conservation: “The habitual diet of prehistoric man would have been low in carbohydrate and high in fat and protein and as such would promote his ability to conserve carbohydrate” (p. 1331).
Our early human ancestors also faced the problem of intermittent access to food, which “would further reinforce this (carbohydrate) economy” (p. 1331).
Albrink asks whether prehistoric man had also developed a “genetic predisposition that favored the storage and ready mobilization of fat and the conservation of body carbohydrate stores. Such a trait would have been of great use to prehistoric man as a hunter and fighter” (p. 1331).
But the “advent of agriculture … brought a change in man’s diet from one composed chiefly of protein and fat to one made up largely of carbohydrate” (p. 1331). Worse, “in recent years the affluence of the Western world had been reflected in the increased availability of highly purified carbohydrate and an increased intake of calories of all kinds” (pp. 1331-1332).
Albrink continued by speculating that if this genotype were to have developed in humans, it would now “carry with it the risk of clinically manifest diabetes mellitus.” She explained:
The common association together of coronary artery disease, obesity, diabetes, and hyper(tri)glyceridemia and their frequent familial occurrence … suggest indeed that all these conditions may be different manifestations of the same genetic trait. Increased insulin secretion and insulin resistance may be additional and perhaps basic manifestations. (p. 1332)
She concluded by saying the articles by Leon Ostrander et al. (47) and Kuo and Bassett (48) “are consistent with the hypothesis that the common modern diseases of diabetes, atherosclerosis, and obesity and associated hyper(tri)glyceridemia may be the present day manifestations of the effect of affluence on a once useful genetic trait, the ability to conserve carbohydrate” (p. 1332).
In the face of such bad news about carbohydrates in general, sugar in particular, Hickson had come to Harvard to enquire whether Hegsted and Stare might be persuaded to go into battle on behalf of his industry. A good place to start would be a favorable review of the literature that would exonerate sugar and rather point the finger of CHD causation squarely at dietary fat. For writing the article, the authors would be paid $500 ($4,100 in today’s dollars). In the end, the total payment to Harvard would balloon to a sizable $6,500 ($52,790 today).
Naturally, to earn their money, the Harvard scientists would be expected to review the literature in a particular way, with a focus on “the several papers which find some special metabolic peril in sucrose and, in particular, fructose” (36, p. 1683). Internal sugar industry documents revealed Hickson’s “particular interest had to do with that part of the nutrition in which there are claims that carbohydrates in the form of sucrose make an inordinate contribution to the metabolic condition, hitherto ascribed to aberrations called fat metabolism. I will be disappointed if this aspect is drowned out in a cascade of review and general interpretation” (p. 1683).
Hegsted assured Hickson that he fully understood the SRF mandate: “We are well aware of your particular interest in carbohydrate and will cover this as well as we can” (p. 1683).
The mission became even more urgent after July 11, 1965, when the New York Herald Tribune featured the Annals articles in a full-page article that included the statement that this new research “threatened to tie the whole business [of diet and heart disease] in a knot” (36, p. 1683). Whereas it had once been thought there was no scientific evidence linking sugar as a cause of coronary atherosclerosis and coronary heart disease, the new research “strengthened the case that sugar increased the risk of heart attack” (36, p. 1683).
Nine months later in April 1966, Hegsted informed the SRF that the project had been delayed by the need to include a rebuttal to the publication of Alfredo Lopez et al. (53). The authors of that study had performed the heinous crime of comparing reported average blood cholesterol concentrations in 16 countries with reported per capita intakes of total calories, fat, simple sugars, complex carbohydrates, and cholesterol. In effect, they were doing with sugar and complex carbohydrate intakes what Keys had done in his original studies for fat and saturated fat (54). The sole difference was that whereas Keys’ graphs would become iconic, that of Lopez et al. (53) has remained hidden — until now (Figure 3).
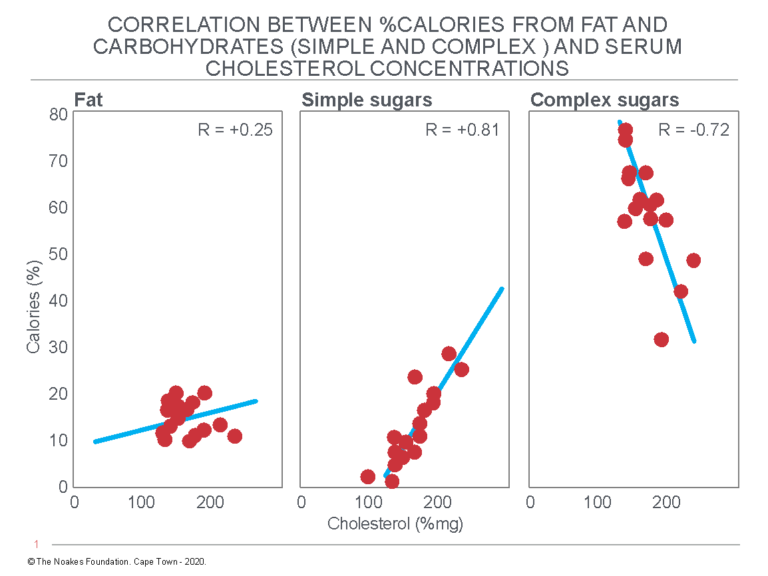
Figure 3: Associational relationships between blood cholesterol concentrations (%mg; mg/dL) and the dietary intakes of fat, simple sugar, and complex sugars, expressed as % of total calories, in 16 countries on four continents. Note the linear relationship between increased intake of simple carbohydrates (sugar) and (rising) blood cholesterol concentrations but the reverse relationship between complex carbohydrates and (falling) blood cholesterol concentrations. Reproduced from Figure 1 in reference 53.
Lopez et al.’s associational data had caused so much distress to the Sugar Research Foundation because it suggested that whereas the consumption of increasing amounts of complex carbohydrates was associated with reduced blood cholesterol concentrations, the increasing consumption of simple carbohydrates — that is, of sugar — produced the opposite effect. In other words, sugar raised blood cholesterol concentrations.
According to Keys’ diet-heart hypothesis, the consumption of sugar would be more likely to cause coronary atherosclerosis than would the consumption of either fat or complex carbohydrates. These data supported Yudkin’s alternate sugar-heart disease hypothesis (54-60) while contradicting Keys’ conclusion that saturated fat is the only dietary factor linked to CHD.
It is understandable why Hickson was so keen to talk to the men from Harvard.
Sixteen months later, by November 1966, two articles written by Robert McGandy, Hegsted, and Stare were ready for submission to the prestigious New England Journal of Medicine (61, 62). One presumes Hickson had been peering eagerly over their shoulders as they wrote these words. Perhaps the SRF even helped in the writing — what we now know as ghostwriting. Either way, Hickson was well pleased with what he read. In a letter to Hegsted he wrote, “Let me assure you this is quite what we had in mind and we look forward to its appearance in print” (36, p. 1684).
Here is what Hegsted and his colleagues conveniently concluded in 1967 in those two published articles (61, 62): “The major evidence today (1967) suggests only one avenue by which diet may affect the development and progression of atherosclerosis. This is by influencing the levels of serum lipids, especially serum cholesterol, though this may take place by means of different biochemical mechanisms not yet understood” (my addition).
“There can be no doubt that levels of serum cholesterol can be substantially modified by manipulation of the fat and cholesterol of the diet,” they wrote, adding:
We conclude, on the basis of epidemiologic, experimental and clinical evidence, that a lowering of the proportion of dietary saturated fatty acids, increasing the proportion of polyunsaturated fatty acids and reducing the level of dietary cholesterol are the dietary changes most likely to be of benefit. The solution here, in our opinion, is a responsibility and opportunity for the food industry — namely, the manufacture of many common foods with characteristics that will lessen the development of atherosclerosis. This is possible today, and only awaits leadership from the food industry. (62, p. 246)
Although the authors did review the inconvenient condition of carbohydrate-sensitive hypertriglyceridemia, they did so only to dismiss it:
Limited evidence from studies on man as well as from researches on laboratory animals show a slightly significant role for the kind and amount of dietary carbohydrate in the regulation of serum lipids. These effects are somewhat more pronounced when diets low in fat are consumed. Since diets low in fat and high in sugar are rarely taken, we conclude that the practical significance of differences in dietary carbohydrate is minimal in comparison to those related to dietary fat and cholesterol. (62, pp. 246-247).
Earlier in the article the authors state: “Until there is more than the currently available meager, direct evidence on the value of dietary management, however, there will inevitably be doubt and controversy” (61, p. 186).
The sole focus in both articles is “healthy” diets that will lower blood cholesterol concentrations on the assumption that any diet producing this effect will also reduce coronary atherosclerosis and protect against CHD. However, at that time, there were no studies to show whether this advice would produce good outcomes. Rather, the men from Harvard simply presumed this outcome was self-evident (because they had decreed it to be so).
Through their articles, the authors managed to convince the world that the sole reason for eating is to lower one’s blood cholesterol concentration. Forgotten, from that moment on, is the simple reality that humans need to ingest multiple nutrients for optimal health.
The authors also ignored all the evidence that an increased carbohydrate intake causes hyper(tri)glyceridemia in proportion to each individual’s degree of insulin resistance — evidence provided by the articles in the June 1965 issue of the Annals of Internal Medicine.
Their explanation that diets low in fat and high in sugar are “rarely taken” is utterly ironic since that is exactly the diet that would be promoted by the 1977 U.S. Department of Agriculture Dietary Guidelines (63). What did these experts expect? Any calories from fat removed from the diet would have to be replaced with calories from carbohydrates, and since fat provides much of the taste in food, sugar would need to be added to fat-free foods to provide a different and more addictive taste.
The authors failed to express any concern that the removal of energy-dense (cholesterol- and saturated-fat-containing) animal products from the diet might produce nutrient deficiencies, especially with regard to certain fat-soluble vitamins.
The authors helped foster the notion that better health would be produced by replacing the naturally occurring foods humans had eaten for millions of years, with factory-produced foods — the products of modern commerce (64). In hindsight, we now know this was an utterly false hope.
In short, the Harvard authors introduced the novel science of dietary reductionism. From that moment the idea gained credence that if a chosen foodstuff lowers the blood cholesterol concentration, then that foodstuff is healthy.
The dominant effect of the two papers by the Harvard authors was essentially to terminate any future discussion about the role of carbohydrates and sugar in the causation of coronary atherosclerosis and CHD. They also managed to relegate the remarkable work of Albrink to a temporary scientific sideshow (50). Her work was largely forgotten and her legacy never properly acknowledged. Instead, the careers of Hegsted, Stare, and other nutrition researchers like Willett, Hu, and Mozaffarian were launched into the stratosphere.
Mid-1960s to Present: The Sugar Industry’s Persuasion of Professor Ian Macdonald
The sugar industry’s ability in the 1960s to make or break academic careers is shown by the manner in which they could “convert” sugar skeptics to their cause. The best example is clearly that of Ian Macdonald.
In 1962 and 1964, Macdonald published two papers (65, 66), the first of which produced results that were confirmed some years later by Kuo and Bassett (38). It showed dietary sucrose ingestion in humans raises blood triglyceride concentrations much more than does the ingestion of the equivalent amount of starch. The second paper (66) found liver fat content increased more in rabbits fed increasing amounts of sucrose or liquid glucose compared to starch (Figure 4).
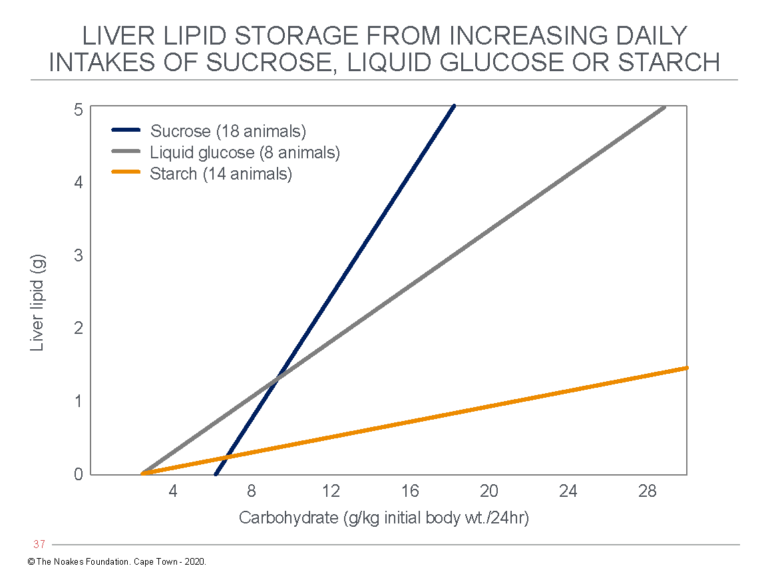
Figure 4: Ian Macdonald’s study found increasing carbohydrate intake from either sucrose, liquid glucose, or starch caused a progressive increase in liver fat (lipid) content in rats, but the increase was the greatest with sucrose ingestion. Reproduced from Figure 1 in reference 66.
Macdonald concluded: “Lipid response in rabbits to dietary carbohydrate is not only related to the level of intake, but also to the type of carbohydrate consumed (66, p. 343). Specifically: “The amount of lipid present in the livers of these animals shows, at any given level of carbohydrate intake, higher values with dietary sucrose than with starch” (66, p. 342).
Clearly these data are very damaging for the sugar industry, particularly today as the incidence of non-alcoholic fatty liver disease (NAFLD) has reached epidemic proportions around the world (67).
Some time between 1964 and the time when he was appointed Professor of Metabolic Physiology, Faculty of Medicine and Health Sciences at the University of Nottingham Medical School, Macdonald seems to have forgotten the grave implications for human health of his original studies. Perhaps it had something to do with his appointment as a member of the European Advisory Boards to Mars and Coca-Cola and, like Willett at Harvard and Mozaffarian at Tufts, because he enjoyed a “strategic relationship” with Unilever (68).
In his new position, he became an international sugar apologist:
But as far as sugar goes, it’s difficult to know where to start because there are people who believe it’s the cause of all of our problems. John Yudkin started this in the 1960s with [his book] Pure, White and Deadly and other people have picked it up at intervals beyond. The consumption rates are a bit higher than they were in the 1960s, but not excessively so. Consumption hasn’t trebled. (68, p. 2)
Actually, how about beginning with the implications for human health of your original studies, Professor?
A subsequent investigation uncovered that between 2012 and 2014, Macdonald’s research unit had received over 1 million pounds from the food industry, including 300,000 pounds from Mars (69).
Perhaps this helps explain why Macdonald’s review articles, like that of the Harvard trio (61, 62), tended to promote the value and safety of dietary carbohydrates. “It is clear that dietary carbohydrates are an essential part of the diet, needed to provide energy for the nervous system” (70, p. 170), he wrote. While there may be concern “on the specific metabolic effects of fructose, which are argued to lead to increased fat deposition in the liver and skeletal muscles with subsequent insulin resistance and increased risk of diabetes” (71, p. S17), there is apparently no need for concern.
He also claimed:
Much of the evidence underpinning these arguments is based on animal studies involving very large intakes of the free sugars. Recent human studies [establish that] … the most marked effects are observed when a high sugar intake is accompanied by an excess energy intake. This does not mean that a high intake of free sugars does not have any detrimental impact on health, but rather that such an effect seems more likely to be a result of the high sugar intake increasing the chances of an excessive intake rather than it leading to a direct detrimental effect on metabolism. (71, p. S17)
Actually no, Professor. It’s not “argued” that this happens. Research findings like your own (Figure 4) have proven it is an established metabolic fact. How quickly you seem to have forgotten.
And no, it’s not correct to blame the effects of sucrose on undisciplined gluttonous humans who simply eat too much. The evidence that sucrose is addictive and that it drives the overconsumption of calories is now well established (72).
Perhaps in time we will learn whether or not Hickson and the SRF played any part in converting Macdonald and others like former University of Cape Town Professor Jim Mann to their cause.
1965-1971: The WHO’s Cooperative Trial in the Primary Prevention of Ischaemic Heart Disease
The first intervention trial (73) to evaluate Keys’ lipid hypothesis was, surprisingly, not conducted in the U.S. but in three European cities: Edinburgh, Budapest, and Prague. The trial became possible with the discovery of the blood-cholesterol-lowering agent chlorophenoxyisobutyrate, later called clofibrate. The drug also lowered blood triglyceride concentrations.
For this trial, 15,745 men aged 30-59 at the start of the trial were randomized into three groups. Half the men whose blood cholesterol concentrations were in the upper third of the blood cholesterol distribution were randomized to receive 1.6 grams of clofibrate daily; the other half with similarly high blood cholesterol concentrations received identical capsules containing olive oil. The third group, with blood cholesterol concentrations within the lowest third of the blood cholesterol distribution, also received the olive oil capsules. This last group was used to compare outcomes in untreated individuals with low blood cholesterol concentrations to those whose blood cholesterol concentrations were equally low as a result of treatment with clofibrate.
The initial hope was that the intervention would produce a 30% reduction in CHD events with a 99% certainty of this result being due only to the drug intervention. The expectation was that this would result from a clofibrate-induced 20% reduction in blood cholesterol concentrations. In the end, clofibrate lowered blood cholesterol concentrations by only 9%.
Despite this disappointing treatment effect, the initial results seemed promising. Although clofibrate did not reduce the number of fatal heart attacks, there were significantly fewer non-fatal heart attacks in those taking the drug. Thus there seemed some hope that cholesterol-lowering might produce beneficial outcomes in CHD prevention.
Unfortunately, there was a problem — a big problem. When the trial ended after five years, there were excess deaths in the clofibrate group: 162 versus 127 in the matched control group. The excess mortality was most likely due to an increased incidence of gallstones in the clofibrate-treated group and the risk of mortality during their surgical removal. In addition, cancer rates — especially of the liver and digestive tract — were also increased in the clofibrate-treated group.
The authors concluded:
The results of the trial confirm the basic hypothesis that reduction of high serum cholesterol levels, even in middle-age, can reduce the incidence of IHD. However, the fact that clofibrate increases the incidence of gallstones, and the possibility that it may have even more serious local pathological consequences, indicate that it cannot be recommended as a lipid-lowering drug for community-wide primary prevention of ischaemic heart disease. (73, p. 1070)
TJ Moore (74) cites the opinion of U.K. epidemiologist Geoffrey Rose (75), who summarized the real challenge posed by such interventions:
In the World Health Organization clofibrate trial (73) … a cholesterol-lowering drug seems to have killed more than it saved, even though the fatal complication rate was only about 1/1000/year. Such low-order risks, which can be vitally important to the balance sheet of mass preventive plans, may be hard or impossible to detect. This makes it important to distinguish two approaches. The first is the restoration of biological normality by the removal of an abnormal exposure (e.g., stopping smoking, controlling air pollution, moderating some of our recently acquired dietary deviations); here there can be some presumption of safety. This is not true for the other kind of preventive approach, which leaves intact the underlying causes of incidence and seeks instead to interpose some new, supposedly protective intervention (e.g., immunization, drugs, jogging). Here the onus is on the activists to produce adequate evidence of safety. (75, p. 432, my emphasis)
The reality is that Rose’s final admonition is seldom applied. The assumption has become that any intervention, be it pharmaceutical or surgical, even if based on unproven theory, is better than doing nothing. The possibility that the intervention could do harm, even if that risk borders on the infinitesimal, is ignored.
The widespread prescription of the modern cholesterol-lowering drugs in the absence of adequate evidence that this practice is either effective or safe (76) is perhaps the most obvious example of how Rose’s injunction has been ignored.
One unexpected outcome of this trial was that it made the senior investigator, Dr. Michael Oliver, a lifelong skeptic of Keys’ diet-heart and lipid hypotheses (77-83). Another prominent British scientist soon joined in (84).
Oliver and John McMichael disagreed vehemently with the relentless drive for consensus among their U.S. colleagues. They later also opposed the desire of the AHA and NHI to enforce interventions on U.S. citizens in the absence of irrefutable evidence that these interventions would do more good than harm.
1966-1986: The NHLBI Initiates the Coronary Drug Project
Between 1966 and 1969, 8,341 U.S. men aged between 30-64 years who had suffered a documented heart attack (myocardial infarction) were recruited by 53 centers participating in the Coronary Drug Project (CDP) (85). The study was designed to evaluate the effects of five different chemical interventions on long-term health outcomes. The interventions tested were the female hormone (equine) estrogen at two different doses, high and low; dextrothyroxine; the vitamin nicotinic acid (niacin); and the drug used in the WHO study (73), clofibrate. All these interventions other than estrogen were chosen because each lowers the blood cholesterol concentration. The study was under the direction of Stamler and Dr. Henry Blackburn, both of whom were staunch allies of Keys.
The initial goal was to study the effects of these interventions for a full five years, but some of the interventions proved problematic already within the first year of the study (86). The estrogen trial was abandoned when it became apparent that giving men the female hormone caused, perhaps predictably, a number of unacceptable side effects, including impotence, reduced sex drive, and breast enlargement.
Dextrothyroxine treatment fared little better, and that trial was terminated when the higher mortality in the treatment group was steadily approaching the threshold for statistical significance.
In the end, only the niacin and clofibrate trials reached completion (87). Although niacin reduced the number of non-fatal heart attacks, total mortality was not reduced by either niacin or clofibrate. In addition, niacin was associated with a number of unacceptable side effects.
With respect to clofibrate, the authors concluded, “The Coronary Drug Project provides no evidence on which to recommend the use of clofibrate in the treatment of coronary heart disease” (87, p. 380).
Similarly for niacin:
The Coronary Drug Project data yield no evidence that niacin influences mortality of survivors of myocardial infarction; this medication may be slightly beneficial in protecting persons to some degree against recurrent non-fatal myocardial infarction. However, because of the excess incidence of arrhythmias, gastrointestinal problems, and abnormal chemistry findings in the niacin group, great care and caution must be exercised if the drug is to be used for treatment of persons with coronary heart disease. (p. 380)
As an aside, it is so interesting that this type of cautionary note, expressed so eloquently in 1975, is essentially never communicated when any of the modern cholesterol-lowering drugs — i.e., statins — are discussed in the medical or popular press. It’s as if caution no longer matters; the public must simply do what they are told, and the medical profession will dictate what the public must believe.
A subsequent 15-year follow-up (88) found mortality in those who had been treated with niacin for five years was 11% lower than in the placebo group. But since those subjects had not taken any niacin for 10 years, it would be illogical to conclude that this effect was due to historical exposure to niacin.
Another long-term analysis found the blood cholesterol concentration was not the best predictor of long-term outcomes in persons who had suffered a previous heart attack. Instead, it was the state of the heart that survived the heart attack that was important: “In these postmyocardial infarction patients, findings indicative of cardiovascular status at baseline evaluation, particularly the state of the myocardium (heart muscle), were more powerful prognosticators of that serum cholesterol” (89, p. 489). As I will discuss subsequently, this finding became even clearer in the five-year follow-up with persons in the placebo arm of the Coronary Drug Project who suffered acute myocardial infarction (heart attack) during the trial (90).
Essentially, the same had been found in Keys’ Seven Countries Study. The presence of Q waves on the electrocardiogram — an indicator of a prior heart attack — was a key predictor of long-term health in that it predicted poorer survival (91, p. 161).
Clearly, this first attempt to improve CHD outcomes by lowering blood cholesterol concentrations had been an abject failure in line with the findings from the WHO clofibrate trial (73).
Both studies gave an early indication that reducing blood cholesterol concentrations might produce unforeseen and unfavorable outcomes.
Perhaps the most important evidence that came out of the Coronary Drug Project arose from an analysis of one subset of men participating in the trial. This finding alone probably justified the conduct of the trial, but the finding, to my knowledge, never has been given appropriate coverage.
A subset of 2,789 patients in the Coronary Drug Project population who developed an acute heart attack (myocardial infarction) and who received the placebo medication were then followed during a five-year period. During that period, 591 of these men died from all causes and the factors associated with a fatal outcome were reviewed.
The key conclusion was, “In general the baseline characteristics that were statistically more related to subsequent mortality were those that reflected the amount of myocardial damage and loss of ventricular function” (90, p. 401). There were 10 such baseline characteristics (p. 401):
- ST-segment depression on the ECG
- Use of diuretics
- New York Heart Association functional class
- Ventricular conduction defects
- Heart rate
- Enlarged heart (cardiomegaly) on chest X-ray
- Number of heart attacks
- History of intermittent claudication (pain in lower limb muscles on walking)
- Serum cholesterol level
- White blood cell count
In other words, the first seven of the 10 factors related to an individual’s state of heart health. One — the presence of intermittent claudication — indicates the extent of atherosclerosis in the lower limb arteries. Another — the white cell count — indirectly reflects the inflammatory state in the body. And only one of the 10 is Keys’ favored blood cholesterol concentration.
When the probability of a fatal outcome was plotted against the prevalence of those 10 characteristics in each individual, the trend was very clear (Figure 5).
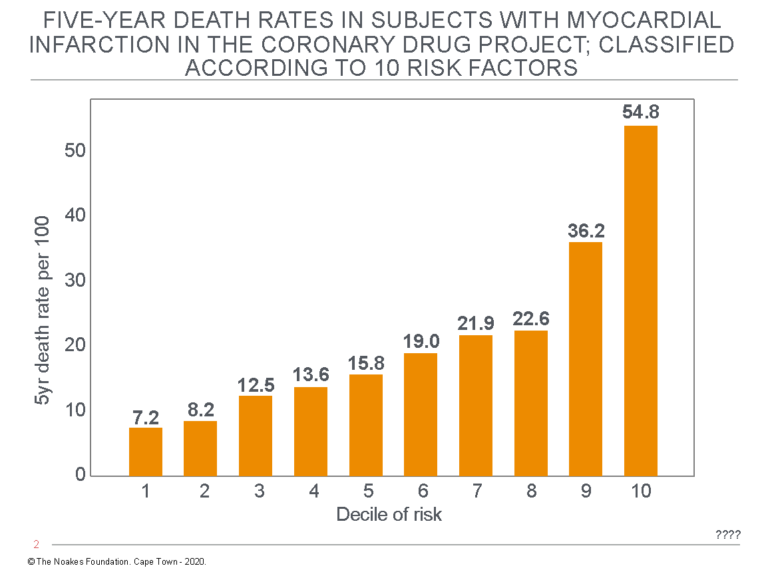
Figure 5: When the subset of subjects in the Coronary Drug Project who suffered a heart attack while receiving the placebo medication during the trial were followed, it was found that their five-year death rate was essentially an exponential function of the extent of their “risk” predicted on the basis of 10 characteristics. Thus, the top 10% of that population with the highest proportion of those 10 risk characteristics (the top decile) were at approximately a seven-fold greater risk than were those in the lowest 10% (the lowest decile). Reproduced from reference 90.
Figure 5 shows that the risk of a fatal outcome during the first five years after a heart attack in this group was essentially an exponential function of the prevalence of the 10 risk characteristics, only one of which was the blood cholesterol concentration.
Thus, the final irony of the Coronary Drug Project was that it was designed to test whether drug-induced lowering of the blood cholesterol reduces subsequent CHD events.
It clearly disproved this hypothesis at least for the drug tested.
What it clearly showed was that mortality in persons with coronary heart disease is best predicted by factors that relate mainly to the degree to which the heart is damaged by the heart attack and the extent of the underlying arterial disease.
Clearly, neither of these is open to modification by reducing the blood cholesterol concentration.
1966-2013: The Sydney Diet Heart Study
Between 1966 and 1973, 458 men between ages 30-59 years who had suffered a recent heart attack and who lived in Sydney, Australia, were randomized into a control group and an intervention group (92). The control group — labeled group P — received “no specific dietary instruction or study foods” (93, p. 1) but were allowed to replace butter with margarine if they so wished (92, p. 319). The intervention group — labeled group F — replaced dietary saturated fat from animal fats, common margarines, and shortenings with omega-6 linoleic acid, a polyunsaturated fatty acid (PUFA), from safflower oil and sunflower oil margarine. Nothing else was changed in either group.
When the data were analyzed and reported in the 1970s (92), the conclusion expressed in the abstract was that the diet had had little effect, either beneficial or harmful. Thus, “none of the dietary factors were significantly related to survival” (p. 317).
Instead, apparently the key factor determining survival was the extent of the coronary and myocardial disease “as judged by clinical parameters” (p. 317). Higher levels of recreational physical activity also “had a strong favorable influence on survival when all other factors were kept constant” (p. 317).
Unfortunately, the abstract was simply untrue — another convenient fib.
The real data showed participants in the P group, which had continued to eat their usual diets, had significantly fewer deaths than those in the F group, which had removed saturated fat from their diets (28 versus 37 deaths, see Figure 6).
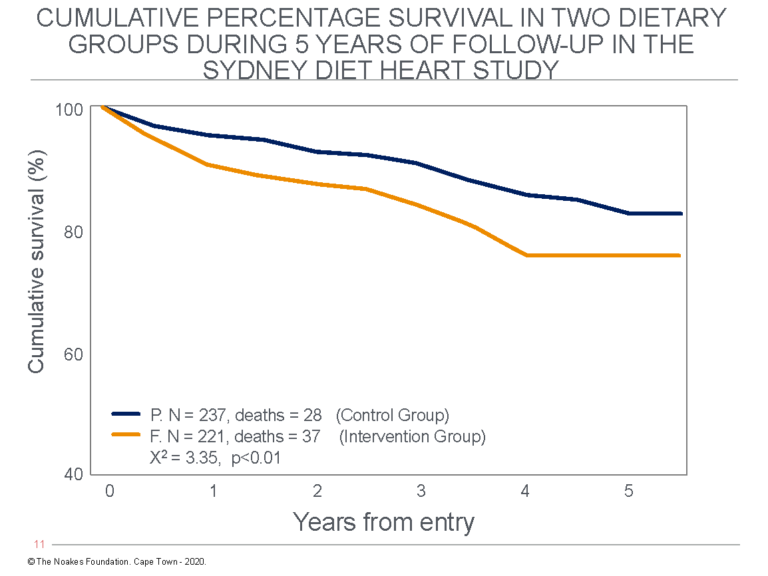
Figure 6: Cumulative percentage survival in the two dietary groups in the original Sydney Diet Heart Study. Note there were significantly fewer deaths and better survival in the control (P) group that continued to eat their normal diets, which included more saturated fats than the intervention group (F) that substituted polyunsaturated for saturated fats. Redrawn from Figure 2 in reference 92 (p. 322).
Naturally, the authors had to explain why they did not find what they had expected. The post-hoc explanation was:
Clearly what was planned as a test of the lipid hypothesis became a multifactorial study. Changes in smoking habit, dietary pattern, body weight, lifestyle and physical activity before and after entry to the trial may well have had a significant effect on prognosis. On that background any added effect of further dietary change could not be shown. (pp. 326-327)
This is of course a nonsense explanation, since there was no evidence the control group had undergone more favorable lifestyle changes than the intervention group.
But the authors did make two other important observations. First, they noted “the lipid hypothesis has gained little support from secondary intervention studies” (92, p. 329). Second, they observed “unselected patients who have had myocardial infarction” have “remarkably good survival … . The annual overall attrition rate of 3 to 4% per annum was similar to that in (other studies)” (92, p. 330).
Contrast this to the modern hysteria that anyone with a slightly elevated blood cholesterol concentration is at risk of dropping dead within days, even if he or she has not suffered a heart attack.
I discuss in a subsequent column (32) what was truly found when these same data were recovered (93) and reanalyzed 40 years later.
1967: Carbohydrate-Sensitive Hyper(tri)glyceridemia Determined to Be the Most Common Lipid Abnormality in Persons With CHD
The previous series on insulin resistance began with the story of how Drs. Albrink and Man had proposed that elevated serum triglyceride rather than higher blood cholesterol concentrations seemed to be the more likely cause of coronary heart disease, especially in those with T2DM (Figures 3-5 in reference 40).
Subsequently, in 1967 Peter Kuo at the Hospital of the University of Pennsylvania reported 82% of persons referred to his lipid laboratory for the investigation of hyperlipidemias, usually hypercholesterolemia, were actually affected by hypertriglyceridemia (50, 94).
Kuo showed the hypertriglyceridemia was caused by high-carbohydrate diets (95), especially those containing sugar (96), since it could be reversed by carbohydrate restriction (Figures 6 and 7 in reference 40; 84).
However, Hickson’s actions on behalf of the Sugar Research Foundation (37, 38), aided by the work of the men from Harvard (61, 62), would essentially bury that work. It remains largely hidden even today.
Additional Reading
- Ancel Keys’ Cholesterol Con, Part 1
- Ancel Keys’ Cholesterol Con, Part 2
- Ancel Keys’ Cholesterol Con, Part 3
- Ancel Keys’ Cholesterol Con, Part 4
- Ancel Keys’ Cholesterol Con, Part 5
References
- Leren P. Prevention of Coronary Heart Disease: Some Results from the Oslo Secondary and Primary Intervention Studies. J Am Coll Nutr 1989;5:407-410.
- Albrink MJ, Lavietes PH, Man EB. Relationship between serum lipids and the vascular complication of diabetes from 1931 to 1961. Trans Assoc Am Physicians 1962;75:235-241.
- Albrink MJ. Triglycerides, lipoproteins, and coronary artery disease. Arch Intern Med 1962;109:345-359.
- Albrink MJ. The significance of serum triglycerides. J Am Diet Assoc 1963;42:29-31.
- Albrink MJ. Diet and cardiovascular disease. J Am Diet Assoc 1965;46:26-29.
- Albrink MJ. Editorial. Carbohydrate metabolism in cardiovascular disease. Ann Intern Med 1965;67:1330-1333.
- Ostrander NS, Kjelsberg MO, Epstein FH. The relationship of cardiovascular disease to hyperglycemia. Ann Intern Med 1965; 62:1188-1198.
- Kuo PT, Bassett DR. Dietary sugar for the production of hyperglyceridemia. Ann Intern Med 1965;62:1199-1212 .
- Noakes TD. Ancel Keys’ cholesterol con, part 6. 13 August 2020. Available here.
- Noakes TD. Ancel Keys’ cholesterol con, part 7. Forthcoming on CrossFit.com.
- Page IH, Stare FJ, Corcoran AC, et al. Atherosclerosis and the fat content of the diet. Circulation 1957;16:163-178.
- Baker BM, Frantz ID, Keys A, et al. The National Diet-Heart Study. An initial report. JAMA 1963;185:141-142.
- Brown HB, Page IH. Highlights of the recent national diet heart study. Clevel Clin Quart 1968;35:131-143.
- Page IH, Brown HB. Editorial. Some observations on the National Diet-Heart Study. Circulation 1968;38:313-315.
- Mojonnier L, Hall Y. The National Diet-Heart Study – assessment of dietary adherence. J Am Diet Assoc 1968;52:288-292.
- Walker WJ. Book Review: The National Diet-Heart Study. Final Report: American Heart Association Monograph 19. Arch Intern Med 1969;123:473-474.
- Teicholz N. The Big Fat Surprise: Why Butter, Meat and Cheese Belong in a Heathy Diet. Simon and Schuster, New York, NY. 2014.
- Central Committee for Medical and Community Program of the American Heart Association. Dietary Fat and its relation to heart attacks and strokes. JAMA 1961;175:389-391.
- Stamler J, Neaton JD. The Multiple Risk Factor Intervention Trial (MRFIT) – Importance then and now. Is Relationship Between Serum Cholesterol and Risk of Premature Death From Coronary Heart Disease Continuous and Graded? Findings in 356 222 Primary Screenees of the Multiple Risk Factor Intervention Trial (MRFIT). JAMA 2008;300:1343-1345.
- Hu FB, Manson JE, Willettt WC. Types of dietary fat and risk of coronary heart disease: A critical review. J Am Coll Nutr 2001;20:5-19.
- Mozaffarian D, Micha R, Wallace S. Effects on coronary heart disease of increasing polyunsaturated fat in place of saturated fat: A systematic review of meta-analysis of randomized controlled trials. Plos Medicine March 23, 2010.
- In declaring his conflicts of interest in the meta-analysis published as reference 11, Mozaffarian lists the following: “Research grants to study the effects of dietary factors on cardiovascular diseases from the US National Institutes of Health; the Searle Scholar Award from the Searle Funds at The Chicago Community Trust; the Genes and Environment Initiative at the Harvard School of Public Health; the Gates Foundation/World Health Organization Global Burden of Diseases, Injuries, and Risk Factors Study; and GlaxoSmithKline, Sigma Tau, and Pronova for an investigator-initiated trial of fish oil to prevent post-surgical arrhythmia. Honoraria and travel expenses for speaking at scientific conferences and reviewing on topics related to diet and cardiovascular disease, including from the U.S. Food and Drug Administration, International Life Sciences Institute, Aramark, Unilever, SPRIM, Nutrition Impact, World Health Organization, UpToDate, and several universities and scientific organizations.”
- Dyer O. International Life Sciences Institute is advocate for food and drink industry, say researchers. BMJ 2019;365:l4037.
- Malkan S. International Life Sciences Institute (ILSI) Is a Food Industry Lobby Group. U.S. Right to Know. Available here.
- Corporate Accountability. Partnerships for an Unhealthy Planet: How big business interferes with global health policy and science. Available here.
- Noakes TD. Ancel Keys’ cholesterol con, part 8. Forthcoming on CrossFit.com.
- But in a more recent (2019) publication: Lee Y, Mozaffarian D, Sy S, et al. Cost-effectiveness of financial incentives for improving diet and health through Medicare and Medicaid: A microsimulation study. PLOS Medicine March 19, 2019. Mozaffarian’s listed conflicts of interest no longer includes reference to Unilever or to the International Life Sciences Institute (ILSI): “DM reports research funding from the National Institutes of Health and the Gates Foundation; personal fees from GOED, Nutrition Impact, Pollock Communications, Bunge, Indigo Agriculture, Amarin, Acasti Pharma, Cleveland Clinic Foundation, America’s Test Kitchen, and Danone; scientific advisory board, Elysium Health (with stock options), Omada Health, and DayTwo; and chapter royalties from UpToDate, all outside the submitted work.”
- Mozaffarian D, Rimm EB, Herrington DM. Dietary fats, carbohydrate, and progression of coronary atherosclerosis in postmenopausal women. Am J Clin Nutr 2004;80:1175-1184.
- Frantz ID, Dawson EA, Ashman PL, et al. Test of effect of lipid lowering by diet on cardiovascular risk. The Minnesota Coronary Survey. Arteriosclerosis 1989;9:129-135.
- Noakes TD. It’s the insulin resistance, stupid: Part 8. CrossFit.com. 24 Nov. 2019. Available here.
- Ramsden CE, Zamora D, Majchrzak-Hong S, et al. Re-evaluation of the traditional diet-heart hypothesis: analysis of recovered data from Minnesota Coronary Experiment (1968-73). BMJ 2016;353:i1246.
- Noakes TD. Ancel Keys’ cholesterol con, part 12. Forthcoming on CrossFit.com.
- Howard BV, Van Horn L, Manson JE et al. Low-fat dietary pattern and risk of cardiovascular disease. The Women’s Health Initiative Randomized Controlled Dietary Modification Trial. JAMA 2006;295:655-666.
- Noakes TD. The Women’s Health Initiative Randomized Controlled Dietary Modification Trial: An inconvenient finding and the diet-heart hypothesis. S Afr Med J 2013;103:824-825.
- Noakes TD. Ancel Keys’ cholesterol con, part 11. Forthcoming on CrossFit.com.
- Kearns CE, Schmidt LA, Glantz SA. Sugar industry and coronary heart disease research. A historical analysis of internal industry documents. JAMA Intern Med 2016;176:1680-1685.
- Kearns C, Schmidt L, Apollonio D, et al. The sugar industry’s influence on policy. Science 2018;360:501.
- O’Connor A. How the sugar industry shifted blame to fat. New York Times. 12 Sept 2018.
- Hegsted DM. Frederick John Stare. Am Soc Nutr Sci 2004;134:1007-1009.
- Hess JL. Harvard’s sugar-pushing nutritionist. The Saturday Review August 1978;10-13.
- Whelan EM, Stare FJ. Panic in the Pantry: Food Facts, Fads and Fallacies. Athenaeum, New York, NY. 1975.
- Epstein FH, Ostrander LD, Johnson BC, et al. Epidemiological studies of cardiovascular disease in a total community – Tecumseh, Michigan. Ann Intern Med 1965;62:1170-1187.
- Nichols AB, Ravenscroft C, Lamphiear DE, et al. Daily nutritional intake and serum lipid levels. The Tecumseh study. Am J Clin Nutr 1976;29:1384-1392.
- Nichols AB, Ravenscroft C, Lamphiear DE, et al. Independence of serum lipid levels and dietary habits. The Tecumseh Study. JAMA 1976;236:1948-1953.
- Noakes TD. Ancel Keys’ cholesterol con, part 5. CrossFit.com. 30 July 2020. Available here.
- Epstein FH. Preventive trials and the “diet-heart” question: Wait for results or act now? Atherosclerosis 1977;26:515-523.
- Ostrander LD, Francis T, Hayner NS, et al. The relationship of cardiovascular disease to hyperglycemia. Ann Intern Med 1965; 62:1188-1198.
- Kuo PT, Bassett DR. Dietary sugar in the production of hyperglyceridemia. Ann Intern Med 1965;62:1199-1212.
- Kuo PT. Hyperglyceridemia in coronary artery disease and its management. JAMA 1967;201:101-108.
- Noakes TD. It’s the insulin resistance, stupid: Part 1. CrossFit.com. 7 July 2019. Available here.
- Albrink MJ. Editorial. Carbohydrate metabolism in cardiovascular disease. Ann Intern Med 1965;67:1330-1333.
- Albrink MJ, Lavietes PH, Man EB. Vascular disease and serum lipids in diabetes mellitus: observations over thirty years (1931-1961). Ann Intern Med 1963;58:305-323.
- Lopez A, Hodges RF, Krehl WA. Some interesting relationships between dietary carbohydrates and serum cholesterol. Am J Clin Nutr 1966;18:149-153.
- Noakes TD. It’s the insulin resistance, stupid: Part 7. CrossFit.com. 11 November 2019. Available here.
- Yudkin J. Diet and coronary thrombosis, hypothesis and fact. Lancet 1957;2;155-162.
- Yudkin J. Nutrition and palatability with special reference to obesity, myocardial infarction, and other diseases of civilisation. Lancet 1963;1;1335-1338.
- Yudkin J. Dietary fat and dietary sugar in relation to ischaemic heart disease ad diabetes. Lancet 1964:2:4-5.
- Yudkin J, Roddy J. Levels of dietary sucrose in patients with occlusive coronary atherosclerotic disease. Lancet 1964;2:6-8.
- Yudkin J. Patterns and trends in carbohydrate consumption and their relation to disease. Proc Nutrition Soc 1964;23:149-162.
- Yudkin J, Morland J. Sugar intake and myocardial infarction. Am J Clin Nutr 1967;20:503-506.
- McGandy RB, Hegsted DM, Stare FJ. Dietary fats, carbohydrates and atherosclerotic vascular disease. N Engl J Med 1967;227:186-192.
- McGandy RB, Hegsted DM, Stare FJ. Dietary fats, carbohydrates and atherosclerotic vascular disease (Concluded). N Engl J Med 1967;227:242-1927.
- Noakes TD. Ancel Keys’ cholesterol con, part 12. Forthcoming on CrossFit.com.
- Price W. Nutrition and Physical Degeneration : A Comparison of Primitive and Modern Diets and Their Effects. Benediction Classics. Garsington, UK. 2010.
- Macdonald I, Braithwaite DM. The influence of dietary carbohydrate on the lipid pattern in serum and in adipose tissue. Clin Sci 1964;27:23-30.
- Macdonald I. Some influences of dietary carbohydrate on liver and depot lipids. J Physiol 1962;162:334-344.
- Fuchs M. Managing the silent epidemic of non-alcoholic fatty liver disease. Fed Pract 2019;36:12-13.
- Boseley S. Sugar intake must come down, says WHO – but UK unlikely to resist. British government’s advisory committee, some of whom receive funding from food industry, sceptical about link with obesity. The Guardian. 7 September 2013.
- Harrison-Dunn A-R. Conflict of interest. On the sugar payroll. Food Navigator.com. 20 January 2014.
- Macdonald IA. Dietary strategies for the management of cardiovascular risk: role of dietary carbohydrates. Nutr Soc 2014;73:167-171.
- Macdonald IA. A review of recent evidence relating to sugars, insulin resistance and diabetes. Eur J Nutr 2016;55(suppl 2):S17-S23.
- Avena MA, Rada P, Hoebel BG. Evidence for sugar addiction. Behavioral and neurochemical effects of intermittent, excessive sugar intake. Neurosci Biobehav Rev 2008;32:20-39.
- Oliver MF, Geizerova H, Gyarfas I, et al. A co-operative trial in the primary prevention of ischaemic heart disease using clofibrate. Report of the Committee of Principal Investigators. Brit Heart J 1978;40:1069-1118.
- Moore TJ. The cholesterol myth. The Atlantic. 1989;264(September):37; Moore TJ. Heart Failure: A Critical Inquiry into American Medicine and the Revolution in Heart Care. New York, NY: Simon and Schuster, 1989.
- Rose G. Sick individuals and sick populations. Int J Epidemiol 2001;30:427-432.
- Diamond D, Ravnskov U. How statistical deception created the appearance that statins are safe and effective in primary and secondary prevention of cardiovascular disease. Expert Rev Clin Pharmacol 2015;8:1-10.
- Oliver M. Dietary cholesterol, plasma cholesterol and coronary heart disease. Br Heart J 1976;38:214-218.
- Oliver MF. Hypercholesterolaemia and coronary heart disease: an answer. BMJ 1984;6415:423-424.
- Oliver MF. Consensus or nonsensus conferences on coronary heart disease. Lancet 1985;1:1087-1089.
- Oliver MF. Prevention of coronary heart disease – propaganda, promises, problems, and prospects. Circulation 1986;73:1-9.
- Oliver MF. Dietary fat and coronary heart disease. Br Heart J 1987;58:423-428.
- Oliver MF. Doubts about preventing coronary heart disease. BMJ 1992;1:393-394.
- Corr LA, Oliver MF. The low fat/low cholesterol diet is ineffective. Eur Heart J 1997;18:18-22.
- McMichael J. Why blame cholesterol? Lancet 1979; 314:1182–1183.
- The Coronary Drug Project Research Group. The Coronary Drug Project. Design, methods and baseline results. Circulation 1973;47:I-1-I-50.
- The Coronary Drug Project Research Group. The Coronary Drug Project. Initial findings leading to modifications of its research protocol. JAMA 1970;214:1303-1313.
- The Coronary Drug Project Research Group. Clofibrate and niacin in coronary heart disease. JAMA 1975;231:360-381.
- Canner PL, Berge KG, Wenger HK, et al. Fifteen year mortality in Coronary Drug Project patients: long-term benefit with niacin. JACC 1986;8:1245-1255.
- Coronary Drug Project Research Group. Natural history of myocardial infarction in the Coronary Drug Project: Long-term prognostic importance of serum lipid levels. Am J Cardiol 1978;42:489-498.
- Schlant RC, Forman S, Stamler J, et al. The natural history of coronary heart disease. Prognostic factors after recovery from myocardial infarction in 2789 men. The 5-year finding of the Coronary Drug Project. Circulation 1982;66:401-414.
- Blackburn H, Menotti A. Major results of the Seven Countries Study. In: Kromhout D, Menotti A, Blackburn H. The Seven Countries Study. A Scientific Adventure in Cardiovascular Disease Epidemiology. Marjan Nijssen-Kramer, Studio RIVM, Bilthoven, The Netherlands. 1993. pp. 159-174.
- Woodhill JM, Palmer AJ, Leelarthaepin B, et al. Low fat, low cholesterol diet in secondary prevention of coronary heart disease. Adv Exper Med Biol 1978; 109:317-31
- Ramsden CE, Zamora D, Leelarthaepin B, et al. Use of dietary linoleic acid for secondary prevention of coronary heart disease and death. Evaluation of recovered data from the Sydney Diet Heart Study and updated meta-analysis. BMJ 2013 Feb 4;346:e8707.
- Kuo PT. Hyperglyceridemia in coronary artery disease and its management. JAMA 1967;201:101-108.
- Kuo PT, Feng L, Cohen NN, et al. Dietary carbohydrates in hyperlipemia (hyperglyceridemia); hepatic and adipose tissue lipogenic activities. Am J Clin Nutr 1967;20:116-125.
- Kuo PT, Bassett DR. Dietary sugar in the production of hyperglyceridemia. Ann Intern Med 1965;62:1199-1212.