Epidemiological and biological evidence have both highlighted the increased incidence of cognitive decline, global brain atrophy, and Alzheimer’s disease (AD) in patients with Type 2 diabetes (T2DM); recent research has sought to understand the common molecular bases connecting these diseases. Whether cause or consequence, inflammation, oxidative stress, amyloid-B accumulation, and mitochondrial dysfunction are present in AD and T2DM and appear to be plausible candidates for the molecular basis linking the two.
Chronic low-grade inflammation is a known component of both obesity and the metabolic syndrome. Macrophages that migrate into fat tissue secrete pro-inflammatory cytokines, such as tumor necrosis factor-𝛼 (TNF-𝛼) and interleukin-6 (IL-6), which have been shown to impair insulin signaling. Neutralizing them improves insulin sensitivity. Other pro-inflammatory cytokines can disrupt the pathways responsible for storing or oxidizing glucose and lipids, which impacts fat accumulation and the handling of sugar and leads to hyperglycemia and dyslipidemia. Hyperglycemia places an increased glucose burden on the beta cells of the pancreas as they work to secrete more insulin to restore normal blood sugar levels. A vicious cycle of inflammation breeding more inflammation ensues, and these inflammatory cytokines are known to be increased in obesity, T2DM, and AD.
A high-carbohydrate diet — the physiological consequence of which will be higher blood glucose, higher insulin secretion, and a greater rate of carbohydrate metabolism — can also result in increased generation of reactive oxygen species (ROS). ROS then can disrupt insulin signaling, bringing oxidative stress into the mix.
In the brain, insulin acts to control appetite, glucose levels, and lipid homeostasis. Insulin receptors are abundant throughout the brain but especially in the hippocampus, where they are known to modulate memory and learning and are necessary for synaptic plasticity. Recent research has shown reduced insulin levels in spinal fluid in the early stages of mild cognitive impairment and AD, and it’s known that reduced glucose metabolism in the brain occurs early in the course of AD. Prolonged high insulin levels reduce the transport of insulin across the protective blood brain barrier by downregulating insulin receptors.
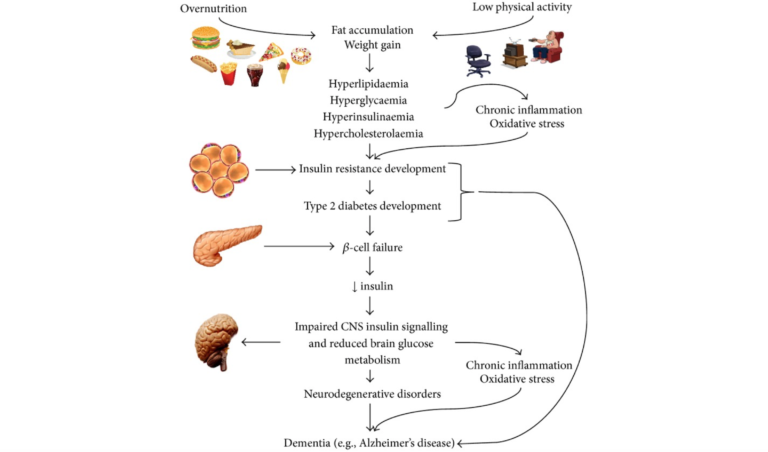
Key hallmarks of AD include brain atrophy (due to neuron cell death), deposition of amyloid plaques, accumulation of intracellular neurofibrillary tangles (NFTs), inflammation, and oxidative stress. The underlying mechanisms by which amyloid accumulates and its downstream events (NFTs and tau protein) develop have yet to be fully explained, but it is known that the accumulated amyloid protein can inhibit autophosphorylation of the insulin receptor and alter a number of cellular processes that result in neuron dysfunction, including insulin signaling. It’s also clear insulin loses some of its effectiveness in the AD brain.
This article’s review of the literature works carefully through common molecular pathways linking disturbed glucose metabolism, oxidative stress, inflammation, and the accumulation of advanced glycation endproducts (AGEs) as they contribute to the progression of both AD and T2DM.