The hypothesis presented here is that injury to the endothelium, by whatever means, leads to the development of a thrombus to cover the damaged area. The thrombus is then covered over by a new layer of endothelium and incorporated into the artery wall before being cleared up by various repair systems.
This process is occurring in all arteries, at all times, in everyone. Normally, this does not cause problems.
However, if the rate of damage exceeds the rate of repair, the damaged area will enlarge, gradually becoming an atherosclerotic plaque. This means factors that can cause plaques to develop and grow do one of three things:
- accelerate endothelial damage
- create larger, more difficult-to-clear thrombi
- impair healing
The glycocalyx and nitric oxide (NO)
There are many factors that can damage the endothelium, some of which are well known, others less so. Before discussing several of them, it is worth looking at two things that protect the endothelium:
- The glycocalyx
- Nitric oxide (NO)
Endothelial cells are covered by a small, dense forest known as the glycocalyx. This is primarily made of strands of sugar(s) and proteins, sometimes known as glycoproteins (or proteo-glycans).
Many fish have scales covered in glycocalyx. This makes them very slippery and difficult to pick up. It also protects the fish by acting as a barrier to the entry of bacteria and suchlike.
The glycocalyx, covering endothelial cells within our blood vessels, has similar functions: It allows the blood to flow freely without sticking, protects the underlying cells from damage, stops blood clots from forming, and prevents entry of potentially damaging substances.
If the glycocalyx is thinned or damaged, this increases the chance of damage to the underlying endothelium. In addition, the glycocalyx contains active enzymes, one of which, nitric oxide synthase (NOS), synthesizes nitric oxide (NO).
Nitric oxide is critical to the health of endothelial cells. It is a vasodilator (widens blood vessels) and thus lowers blood pressure. It is also a potent anticoagulant (stops thrombi from forming), prevents adhesion of platelets and white blood cells, and also stimulates the formation of endothelial progenitor cells (EPCs) in the bone marrow.
If the glycocalyx is healthy and there is sufficient production of NO, then the underlying endothelial cells will be protected.
Factors that can accelerate damage to the glycocalyx/endothelium
Many factors can damage the endothelium. Some of the more common are:
- Smoking
- Raised blood sugar
- Air pollution
- Raised blood pressure
- Bacterial infections
- Raised stress hormone levels (e.g., cortisol)
- Steroids (corticosteroids)
Let’s look at one of these in more detail: smoking. Studies have demonstrated that smoking a single cigarette damages the endothelium (as measured by looking at microparticles (MPs), which can be detected in the blood after endothelial cells die).
In parallel with the damage, the bone marrow will produce more endothelial progenitor cells (EPCs) to cover the area of damage that has resulted.
As F. Mobarrez et al. explain, “Brief active smoking of one cigarette generated an acute release of EPC and MPs, of which the latter contained nuclear matter. Together, these results demonstrate acute effects of cigarette smoke on endothelial, platelet and leukocyte (white blood cell) function as well as injury to the vascular wall” (my emphasis).
Less common but very damaging factors include diseases associated with severe vasculitis. Vasculitis is a condition characterized by acute inflammation and damage to the artery wall/endothelium, often due to an autoimmune reaction such as:
- Kawasaki disease
- Rheumatoid arthritis
- Systemic lupus erythematosus
- Scleroderma
- Sjögren’s syndrome
It is possible for children as young as four or five to die from CVD, having suffered an acute episode of Kawasaki disease. Systemic lupus erythematosus can increase the risk of CVD death by 5,000% in younger women. Any condition that causes vasculitis increases CVD risk.
Aside from vasculitis, the disease that may cause the most rapid and severe endothelial damage is sickle cell disease (SCD). Here, sharp and rigid (sickled) red blood cells (RBCs) can create intense physical disruption to the glycocalyx and endothelial cells, thus driving atherosclerosis and CVD death.
Below is a short excerpt from a case history of a 14-year-old boy with SCD. He had gangrene in his foot due to significantly reduced blood supply (ischaemia), caused by severe atherosclerosis. He also had calcified atherosclerosis in all the main arteries in his body, which is normally a late stage of atherosclerotic plaque development only seen in older adults:
A 14 year-old boy was referred to our vascular unit, with gangrene of the right foot. The condition started about 1 year prior to this referral with ulceration of the foot which was treated conservatively. The condition of the foot deteriorated until development of gangrene of most of the foot. The boy is a known patient of SCD. His past medical history revealed right sided stroke when he was 8 years old. His parents have SCD. His brother had also SCD and died suddenly at the age of 5 years.
The boy had no established risk factors for CVD. SCD provides compelling evidence that damage to the endothelium is the primary driver for the development of atherosclerotic plaques.
Creating larger and more difficult-to-repair thrombi
There are a few procoagulant conditions, such as Hughes syndrome, or antiphospholipid syndrome (APS). Here, the blood is hypercoagulable, and people with this syndrome develop atherosclerosis at a faster rate and are far more likely to die from CVD. The progression of CVD can be controlled, to an extent, with anticoagulants such as aspirin.
It has also been found that people who have higher levels of fibrinogen, which forms fibrin, a key component of blood clots, are more likely to die from CVD. On the other hand, people with hemophilia (blood less likely to clot) are significantly less likely to die from CVD.
One factor that makes any blood clot that forms far more difficult to lyse (break down) is lipoprotein A (Lp(a)). Lp(a) is low-density lipoprotein (LDL) with an additional protein attached to it: apolipoprotein A (ApoA). This protein, attracted to areas of endothelial damage, forms strong bonds with the exposed arterial wall.
ApoA also blocks clot lysis. It mimics plasminogen, a protein incorporated into all thrombi as they form. Plasminogen is, in turn, converted to plasmin by tissue plasminogen activator (TPa). Plasmin lyses strands of fibrin, thus “busting” the clot (fibrinolysis). TPa, manufactured as a drug, is known as a clot buster and is used to treat strokes and heart attacks. If there is a higher concentration of Lp(a) in a thrombus, the action of TPa will be inhibited.
The attraction of Lp(a) to areas of damage, and the difficulty in breaking down the surrounding clot, is probably why Lp(a) can be found in high concentrations within atherosclerotic plaques. It also raises the possibility that much or all the LDL found within plaques may be misclassified Lp(a). The two lipoproteins are identical, other than the ApoA, and unless this is specifically looked for, it will not be noted.
Impaired healing
For damage to occur at a faster rate than healing or repair, damage must be accelerated or healing impaired — or both. It is worth looking at factors that can impair healing after endothelial damage. Two extreme examples are immunosuppressants and vascular endothelial growth factor inhibitors.
Immunosuppressants are used for a variety of different diseases. However, their use in preventing organ rejection is of interest here. Immunosuppression is, essentially, a dampening down of the healing processes in the body. This can be a good thing if you are trying to prevent rejection of a new organ.
However, it can also be harmful. Steroids were one of the first immunosuppressants, used in many conditions to dampen down inflammation, but their use also severely impacts the healing process. Steroids significantly increase the risk of CVD.
One of the major problems after organ transplantation is graft allograft vasculopathy (AV). At its most simple, AV can be viewed as very rapid development of atherosclerosis.
As E. Tepperman et al. note, “Many of the currently used immunosuppressants cause endothelial dysfunction after transplantation and may further accelerate the development of intimal hyperplasia and AV.”
Vascular endothelial growth inhibitors (VEGF inhibitors) are used in cancer treatment, primarily to suppress angiogenesis (the development of new blood vessels that can feed the tumor). They are effective. However, as a side effect of their central function, they significantly impair endothelial growth and repair.
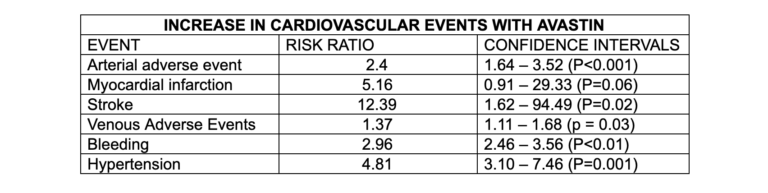
A meta-analysis of the use of Avastin (a widely used VEGF inhibitor) found it significantly increased the risk of atherosclerosis and CVD death.
Summary
Atherosclerotic plaques start and grow when damage to the artery wall/endothelium exceeds the capacity of the repair processes. This means factors that can cause CVD do one of three things:
- Accelerate damage to the endothelium
- Create larger and more difficult-to-heal thrombi
- Impair the healing processes
Viewing CVD in this way links risk factors for the disease that may not seem to have anything in common — for example, sickle cell disease and Avastin, or smoking and immunosuppressants, or air pollution and rheumatoid arthritis, or bacterial infections and Sjögren’s syndrome, or Hughes syndrome and a raised blood sugar level.
The next article will review some of the most effective ways of protecting the endothelium, improving repair processes, and controlling blood coagulability.
Additional Reading
- What Causes Cardiovascular Disease? The Response to Injury Hypothesis, Part 1
- What Causes Cardiovascular Disease? The Response to Injury Hypothesis, Part 2
 Malcolm Kendrick is a family practitioner working near Manchester in England. He has a special interest in cardiovascular disease, what causes it, and what may prevent it. He has written three books: The Great Cholesterol Con, Doctoring Data, and A Statin Nation. He has authored several papers in this area and lectures on the subject around the world. He also has a blog, drmalcolmkendrick.org, which stimulates lively debate on a number of different areas of medicine, mainly heart disease.
Malcolm Kendrick is a family practitioner working near Manchester in England. He has a special interest in cardiovascular disease, what causes it, and what may prevent it. He has written three books: The Great Cholesterol Con, Doctoring Data, and A Statin Nation. He has authored several papers in this area and lectures on the subject around the world. He also has a blog, drmalcolmkendrick.org, which stimulates lively debate on a number of different areas of medicine, mainly heart disease.
He is a member of THINCS (The International Network of Cholesterol Sceptics), which is a network of doctors and scientists who believe that cholesterol is not the main underlying cause of heart disease. He remains a proud Scotsman, whisky drinker, and failed fitness fanatic who loves a good scientific debate — in the bar.