For the past 170 years, there have been two main competing ideas about what causes cardiovascular disease (CVD). One of them, the cholesterol hypothesis, has come to dominate. The other is far less known.
The cholesterol hypothesis, in its simplest form, states increased blood cholesterol is deposited in the artery wall, leading to thickening and narrowing in the arteries. This is known as atherosclerosis or atherosclerotic plaques.
The alternative hypothesis has had different names over the years — e.g., the encrustation hypothesis, thrombogenic hypothesis, and response to injury hypothesis. Whatever name is given, this hypothesis, in its simplest form, suggests damage to the lining of the artery causes blood clots to form on the artery wall. These blood clots are then incorporated into the artery, growing into atherosclerotic plaques.
There is little overlap between these two competing hypotheses. If one is correct, then the other cannot be. Therefore, the question here is: Can the response to injury hypothesis explain most or all the facts known about heart disease? In this four-article series, I hope to explain more about this hypothesis and how it works.
A brief history of the response to injury hypothesis
As Russell Ross et al. note, “In 1856, Rudolf Virchow proposed that the lesions of atherosclerosis result from injury to the artery wall” (1).
At around the same time period, another famous researcher and pathologist named Karl von Rokitansky suggested atherosclerotic plaques were blood clots in various stages of repair. He called this the “encrustation” hypothesis, and he ascribed all the features of atherosclerosis to “recurrent deposition of elements derived from the blood mass.” Essentially, when he studied atherosclerotic plaques, he believed he was looking at thrombi.
Unfortunately, Rokitansky and Virchow disagreed. Virchow noted plaques lay underneath the inner layer of the artery wall, the endothelial layer (or endothelium). He did not believe blood clots could possibly develop anywhere other than within the bloodstream. This criticism was taken up by others, and Rokitansky’s hypothesis fell out of mainstream thinking.
However, CVD was still a relatively uncommon condition at that time. It did not emerge as the most common cause of death until the mid-20th century. This meant research and discussion on the causes and processes associated with CVD did not really progress a great deal for nearly a hundred years.
It was only when the “epidemic” of CVD struck the U.S. and U.K. after the Second World War that interest in CVD sharpened. Almost immediately, versions of the response to injury hypothesis were resurrected, primarily by a researcher called J. B. Duguid.
As Neville Woolf writes:
Much of the comparatively recent interest in the role of mural thrombi (blood clots developing on the surface of artery wall) as a contributor to plaque growth stems from the studies of J. B. Duguid, who stated that many of the lesions we now classify as atherosclerosis are arterial thrombi which, by the ordinary processes of organization, have been transformed into fibrous thickenings. This view as Duguid pointed out represents a partial return to the “encrustation hypothesis” enunciated by Rokitansky. (2, my addition)
As with Rokitansky, although Duguid’s ideas gained attention, the alternative cholesterol hypothesis was taken up with far greater enthusiasm.
Still, the response to injury hypothesis did not die out completely. It was resurrected again by Ross in the 1970s. In 1977, in a paper called “Response to injury and atherogenesis,” he wrote:
This hypothesis states that the lesions of atherosclerosis result as a response to some form of injury to arterial endothelial cells that result in their desquamation (damage/stripping off). The injury may be subtle. There may be many potential sources of injury; these include chronic hyperlipidaemia, various chemical factors such as homocystine, uremia, metabolites, infections, immunologic injury, and mechanical factors. Mechanical injury may occur at particular anatomic sites as a result of the increased shear stress applied to the endothelial cells from the flow of the blood at these sites. (1, my addition)
As he went on conclude:
The studies reported here support the theme that atherosclerosis results from injury to the endothelium followed by endothelial desquamation and platelet adherence, aggregation, and release at the sites of exposed subendothelial connective tissue. (1)
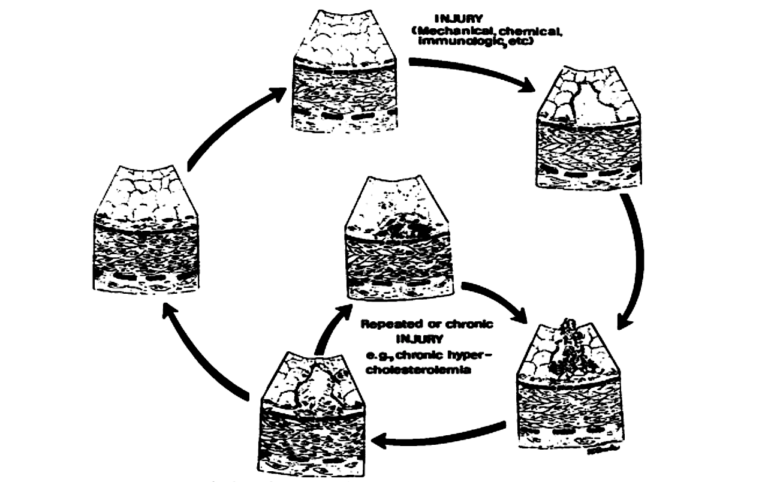
Figure 1: Response to injury hypothesis (reproduced from reference 1)
For some reason, Ross did not describe platelet adherence and aggregation as a thrombus although that is, essentially, what a thrombus consists of when it first forms. Perhaps he was aware of Virchow’s criticism of Rokitansky and did not wish to claim he was looking at thrombi lying within the artery wall.
He was also trying to incorporate the cholesterol hypothesis within the response to injury hypothesis by including hyperlipidaemia as a cause of arterial injury. Hyperlipidaemia is, essentially, a different name for a high blood cholesterol level.
As before, the idea gained a certain following and interest but never really developed enough momentum to enter mainstream thinking. Perhaps the last researcher to resurrect the response to injury hypothesis was Elspeth Smith in the 1980s. As she and W. Douglas Thompson wrote:
After many years of neglect, the role of thrombosis in myocardial infarction is being reassessed. It is increasingly clear that all aspects of the haemostatic system are involved: not only in the acute occlusive event, but also in all stages of atherosclerotic plaque development from the initiation of atherogenesis to the expansion and growth of large plaques. (3)
Around this time, the first statins were launched into the market. They lowered blood cholesterol by around 30 to 40%, purported to reduce the risk of CVD, became the most profitable medications in the history of medicine, and drew a line over any research into CVD that did not place the cholesterol hypothesis at the center — at which point, the response to injury hypothesis became an idea confined to history.
However, the response to injury hypothesis is almost certainly capable of explaining the pattern of CVD and CVD risk far more accurately than the cholesterol hypothesis. The next article will look at the main objections to the response to injury hypothesis and why it has not yet managed to become dominant.
 Malcolm Kendrick is a family practitioner working near Manchester in England. He has a special interest in cardiovascular disease, what causes it, and what may prevent it. He has written three books: The Great Cholesterol Con, Doctoring Data, and A Statin Nation. He has authored several papers in this area and lectures on the subject around the world. He also has a blog, drmalcolmkendrick.org, which stimulates lively debate on a number of different areas of medicine, mainly heart disease.
Malcolm Kendrick is a family practitioner working near Manchester in England. He has a special interest in cardiovascular disease, what causes it, and what may prevent it. He has written three books: The Great Cholesterol Con, Doctoring Data, and A Statin Nation. He has authored several papers in this area and lectures on the subject around the world. He also has a blog, drmalcolmkendrick.org, which stimulates lively debate on a number of different areas of medicine, mainly heart disease.
He is a member of THINCS (The International Network of Cholesterol Sceptics), which is a network of doctors and scientists who believe that cholesterol is not the main underlying cause of heart disease. He remains a proud Scotsman, whisky drinker, and failed fitness fanatic who loves a good scientific debate — in the bar.
References
- Ross R, et al. Response to injury and atherogenesis. Am J Pathol. 86.3(March 1977): 675-84. Available here.
- Woolf N. Thrombosis and atherosclerosis. Brit Med Bull. 34.2(May 1978): 137–142. Available here.
- Smith EB, WD Thompson. Fibrin as a factor in atherogenesis. Thrombosis Research 73(1994): 1-19. Available here.