Question
What role does insulin resistance in skeletal muscle play in the development of fatty liver disease, obesity, and other elements of the metabolic syndrome?
Takeaway
This small study indicates skeletal muscle insulin resistance may be the primary (initial) defect in metabolic deterioration. Insulin-resistant subjects, compared to matched, insulin-sensitive subjects, shuttled a larger share of ingested calories toward triglyceride production than glycogen storage. This shift, which occurred in subjects with normal weight and inflammation levels, would be expected to lead to increased liver and visceral fat storage, hepatic and peripheral insulin resistance, adiposity, and the other features of the metabolic syndrome. These results thus suggest muscular insulin resistance is the primary defect in the metabolic syndrome in at least some subjects, and further, that interventions to preserve or promote muscular insulin sensitivity may be particularly effective at preventing or reversing the metabolic syndrome.
In this 2007 study, 12 young, healthy, insulin-resistant men (1) were compared to 12 insulin-sensitive but otherwise similar men. Inflammation levels, weight, fat mass, and activity levels were similar between groups. The insulin-resistant subjects had lower baseline HDL, higher uric acid levels, and higher fasting plasma glucose, insulin, and triglyceride levels.
Both groups were given a high-carbohydrate meal (2). The subjects in both groups showed similar postprandial increases in blood glucose levels, while triglyceride and insulin levels increased more in insulin-resistant subjects than in their insulin-sensitive counterparts. Using labeled carbohydrates, researchers were able to track the fate of the ingested carbs. In the insulin-resistant subjects, muscle glycogen synthesis was 61% lower than in insulin-sensitive subjects; hepatic triglyceride synthesis and hepatic DNL were 2.5 and 2.2 times higher, respectively. In other words, a larger share of the ingested carbohydrates were shuttled toward triglyceride production rather than being stored as glycogen in insulin-resistant subjects.
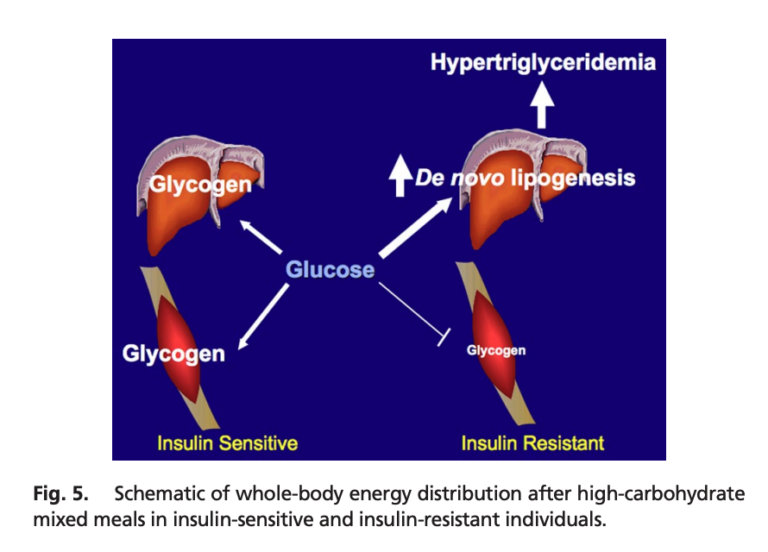
Increased triglyceride production in insulin-resistant subjects accordingly led to higher levels of postprandial VLDL and lower levels of postprandial HDL than in insulin-sensitive subjects. This pattern is consistent with previous research indicating skeletal muscle insulin resistance leads to increased plasma triglycerides and reduced plasma HDL (3).
This study illustrates the primary role muscular insulin resistance can play in the development of metabolic disease. As shown here, muscular insulin resistance — observed prior to any defects in overall adiposity or inflammation — leads to a greater share of consumed calories being directed toward triglyceride production and a smaller share toward glycogen storage, even when identical meals are consumed. This increased triglyceride production would be expected to lead to fatty liver disease and hepatic insulin resistance (4), which itself leads to fasting hyperglycemia, obesity, and diabetes (5). The fact that these defects were visible in subjects with normal body weight and normal levels of inflammation suggests such factors are secondary defects and impaired muscular insulin sensitivity is the initial cause of the metabolic syndrome in at least some individuals (6). This suggests interventions to maintain and improve the ability of muscle cells to take up glucose in response to insulin — including, as previously reviewed on CrossFit.com, exercise — may play a particularly important role in the prevention and/or amelioration of the metabolic syndrome.