What Professor Paul Sonksen calls the “‘black age’ of endocrinology” set in after researchers in the late 1950s published work revealing insulin stimulated the uptake of glucose by muscle cells. These experiments, done in vitro on isolated bits of rat muscle, were erroneously extrapolated to describe the pathophysiology of diabetes in living humans. At the time, the assumption was that hyperglycemia was driven by a lack of glucose disposal. In other words, sugar levels in the blood rose, because there was no (or insufficient) insulin to drive glucose into the cells. From this incorrect assumption, the idea arose that glucose was dependent upon insulin to get into the cells, and that exogenous insulin reduces hyperglycemia by driving glucose into the cells, particularly the muscle cells.
Although work in the 1970s disproved this theory, it has persisted. In fact, it has been taught that way for so long, it has become dogma. As Sonksen points out, “Even in well respected texts it is still common to find statements such as ‘The basic action of insulin is to facilitate glucose entry into cells, primarily skeletal muscle and hepatocytes’” (1). In discussions with multiple physicians, all remember similar instruction in medical school and continue to operate under that same false assumption.
There are plenty of glucose transporters in the cell membrane in the fasting state to facilitate glucose transport into the cells in the face of an absolute insulin deficiency as long as there exists a sufficiently high glucose gradient. There are more transporters in the cytoplasm that insulin can (and does) mobilize and bring to the cell membrane, but glucose uptake is never totally insulin dependent in humans — even in states of severe ketoacidosis.
These facts would seem to fly in the face of the reality that insulin injections do indeed quickly bring down elevated blood glucose levels in diabetic patients. But the mechanism is not that it increases glucose uptake by the cells — that is already underway. The injected insulin inhibits the liver’s output of glucose via gluconeogenesis. In diabetic patients, the liver produces and releases glucose at a much higher rate than the cells can absorb it, running blood levels ever higher. Exogenous insulin shuts off the faucet; it does not open the drain. The drain is already open. In fact, as Paul Sonksen and J. Sonksen show, exogenous insulin can actually reduce the uptake of glucose under hyperglycemic conditions (1).
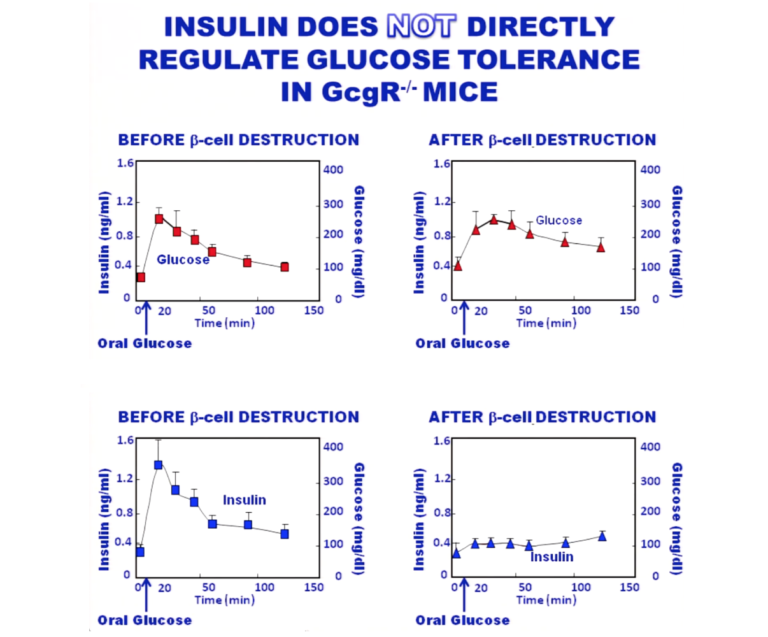
Work by Y. Lee et al. shows that in glucagon receptor knockout mice, glucose tolerance curves are essentially the same in mice with and without functional beta cells — i.e., with and without endogenous insulin (2). These animals have no functional glucagon receptors, so there is no influence of glucagon on the curves. The top graphic in the figure below shows the glucose tolerance curve in both the presence and absence of insulin. The bottom shows that insulin has been successfully eradicated in the animals, so there is no insulin effect present.
Sonksen and Sonksen detail the work done to elucidate the actions described above. They also describe the multiple actions of insulin in its inhibition of lipolysis along with its actions on glucose production and storage. This classic paper serves as an excellent primer on all aspects of glucose action, ketosis, lipolysis, and the many ways diabetes can manifest. It is well worth the time taken to study.
